Das Potenzial von Routineanalysen des mikrobiellen CF-Atemwegsmetagenoms
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Bakterielle Pathogene spielen bei der Besiedlung der Atemwege eine nicht zu vernachlässigende Rolle. Eine stark reduzierte bakterielle Diversität bei gleichzeitig erhöhter Präsenz eines oder mehrerer Pathogene gilt als typisch für das CF-Lungenmikrobiom. Routinemäßige Atemwegsmetagenomanalysen sowie die Festlegung von Grenzwerten könnten bei CF-Patienten erheblich zum Behandlungserfolg beitragen.
Keypoints
Sequenzierverfahren ermöglichen den schnellen Nachweis von abundanten und seltenen Bakterien und ihrer Antibiotikaresistenzdeterminanten aus einer Patientenprobe.
Das longitudinale Tracking pathogenspezifischer DNS könnte dazu beitragen, bakterielle Atemwegsinfektionen und die Entstehung einer irreversiblen CF-Mikrobiomsignatur zu verhindern.
Die Evaluierung der Wachstumsdynamik einer Erregerpopulation in den Atemwegen ermöglicht es, die Teilungsrate der Bakterien kontinuierlich zu überwachen.
Fehlende Standardrichtlinien verhindern aktuell die Einführung von Routineanalysen des CF-Atemwegsmetagenoms in die klinische Diagnostik.
Obwohl die Mukoviszidose (bzw. zystische Fibrose, CF) eine Multisystemerkrankung ist, lassen sich die erhöhte Morbidität und vorzeitige Mortalität der Patienten primär durch chronische Infektionen und Entzündungen der unteren Atemwege erklären. Die bakteriellen Pathogene und ihre Antibiotikaresistenzdeterminanten werden routinemäßig über kulturabhängige Verfahren bestimmt. Da zwischen Probenahme und Diagnostik oftmals 48–72 Stunden liegen, kann sich der Therapiebeginn mit erregerspezifischen Antibiotika verzögern.1 Währenddessen destabilisiert die empirische Breitbandantibiotikatherapie unter Umständen das gesamte Lungenmikrobiom und begünstigt ein konkurrenzloses Wachstum von resistenten Keimen.2 Außerdem muss die Bakterienlast des Erregers mindestens 103–105c.f.u./ml1 betragen, damit dieser erfolgreich im Kulturmedium der Mikrobiologie anwächst.1 Das Auftreten dieses Lungenmikrobiommusters mit stark reduzierter Bakteriendiversität und der erhöhten Präsenz einer oder mehrerer Pathogene wurde in der Literatur als CF-typisch und als irreversible Mikrobiomsignatur im fortgeschrittenen Krankheitsverlauf beschrieben.3
Die Atemwegsmetagenomik
In der Atemwegsmetagenomik wird die gesamte in der Patientenprobe befindliche humane und mikrobielle Erbsubstanz isoliert und sequenziert, z.B. aus dem tiefen Rachenabstrich, der bronchoalveolären Lavage oder dem induzierten Sputum. Die DNS-Fragmente werden daraufhin gegen eine Datenbank kartiert, welche die Referenzsequenzen der humanen Chromosomen, Bakterien, DNS-Viren, Phagen und Pilzen enthält. Mit dem Verfahren der sensitiven Hochdurchsatzsequenzierung können innerhalb weniger Stunden Mikroorganismen aus einer einzigen Patientenprobe taxonomisch bestimmt werden.1,3,4 Es ist zusätzlich möglich, die Virulenz- und Antibiotikaresistenz-assoziierten Gene der Mikroorganismen in der Patientenprobe zu analysieren.1,5
Im Rahmen einer kürzlich durchgeführten longitudinalen Atemwegsmetagenomstudie konnten wir zeigen, dass geringe DNS-Mengen des CF-Erregers Pseudomonas aeruginosa in den unteren Atemwegen von Neugeborenen und Kleinkindern mit CF vorhanden sein können, bevor der Erreger in der Kultur nachweisbar ist. Dabei waren die folgenden drei P.-aeruginosa-Erscheinungsmuster im Kindesalter erkennbar (Abb.1):
-
Abb. 1A: Der klassische CF-Modus (selten im Kleinkindalter) mit akuter und dann chronischer Besiedlung der unteren Atemwege, nachgewiesen über das kulturunabhängige Sequenzierverfahren und die kulturabhängige Diagnostik
-
Abb. 1B: Kein Nachweis von P.-aeruginosa-spezifischer DNS
-
Abb. 1C: Der stochastische Nachweis von P.-aeruginosa-spezifischer DNS ohne Anhaltspunkte für eine bakterielle Persistenz in den Atemwegen. In diesem Fall wurde P.-aeruginosa ausschließlich über das Sequenzierverfahren detektiert.
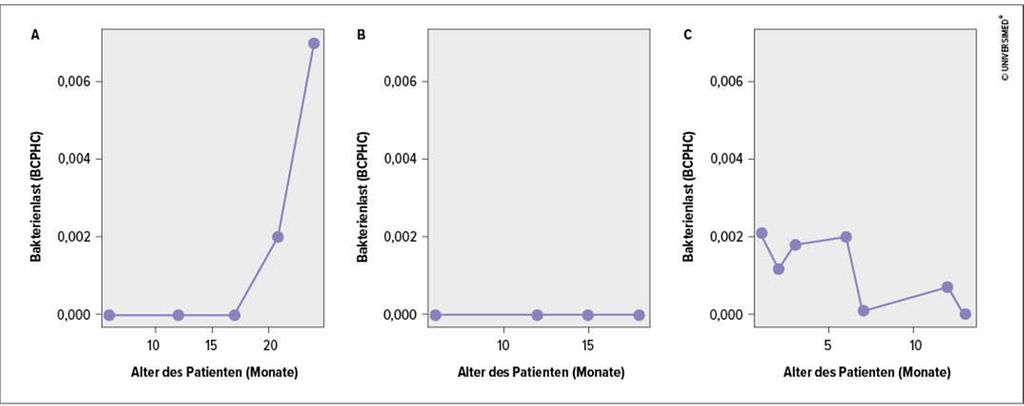
Abb. 1: Die drei P.-aeruginosa-Erscheinungsmuster im Kindesalter. (A) Der klassische CF-Modus mit pathogener Besiedlung der unteren Atemwege. (B) Kein Nachweis von P.-aeruginosa-spezifischer DNS. (C) Der stochastische Nachweis von P.-aeruginosa-spezifischer DNS ohne Anhaltspunkte für eine bakterielle Persistenz in den Atemwegen. BCPHC: „bacterial cell per human cell“3
Interessanterweise war das P.-aeruginosa-Erscheinungsmuster3 (Abb.1C) nichtkrankheitsassoziiert. Auch bei gesunden Kleinkindern ließ sich der Erreger stochastisch mit ähnlicher Detektionsrate nachweisen. Da P.-aeruginosa in der Umwelt weit verbreitet ist, gehen wir davon aus, dass auch gesunde Kleinkinder regelmäßig in den Kontakt mit geringen Mengen des Erregers kommen und es eventuell von Vorteil sein könnte, um eine spezifische Immunantwort auszubilden, die vor späteren Atemwegsinfektionen mit P.-aeruginosa schützt. Ein routinemäßiges Screening des Lungenmetagenoms mit einem longitudinalen Tracking pathogenspezifischer DNS könnte dazu beitragen, bakterielle Atemwegsinfektionen zu verhindern und die Entstehung einer irreversiblen CF-Mikrobiomsignatur im weiteren Krankheitsverlauf zu vermeiden.
Berechnung der Wachstumsdynamik einer Erregerpopulation
Neben der taxonomischen Bestimmung der Mikroorganismen in einer Probe lassen sich weitere wichtige Informationen aus den Sequenzierdaten herauslesen. Die am häufigsten vorkommenden Bakterien der Atemwege haben ein zirkuläres Genom und teilen sich bidirektional; die Replikation beginnt am Replikationsursprung und endet dort, wo die beiden Replikationsgabeln aufeinandertreffen (Terminus). In der exponentiellen Wachstumsphase starten Replikationszyklen, bevor die vorhergegangenen abgeschlossen sind. Sequenziert man die DNS einer sich schnell teilenden Erregerpopulation und kartiert die Fragmentstücke gegen das dazugehörige Referenzgenom, so erhält man einen Gradienten aus Fragmentstücken entlang des Genoms mit einer erhöhten Anzahl von Fragmenten am Replikationsursprung und einer reduzierten Anzahl am Terminus.6 Das Ausmaß des Gradienten kann quantitativ bestimmt werden, um die Wachstumsdynamik der Erregerpopulation bzw. die Teilungsrate des Pathogens in der mikrobiellen Lebensgemeinschaft zu berechnen. Routineanalysen des CF-Atemwegsmetagenoms würden es ermöglichen, Pathogene mit reduzierter Aktivität in der CF-Lunge von Pathogenen in der frühen oder späten exponentiellen Wachstumsphase zu unterscheiden. Außerdem könnte der Effekt von Umwelteinflüssen auf die Erregerpopulation longitudinal evaluiert werden, wie zum Beispiel der Beginn einer medizinischen Intervention oder ein Therapiewechsel.
Limitationen
Die Qualität und die Reproduzierbarkeit der Sequenzierergebnisse werden sowohl von dem Probentyp, der Qualität der Probenahme, dem ausgewählten Labor- und Bioinformatikverfahren als auch von der Sauberkeit der Klinik- und Laborumgebung beeinflusst.7,8 Bisher gibt es keine Standardrichtlinien für die Probenaufarbeitung, Sequenzierverfahren oder die zu nutzenden Referenzdatenbanken. Ebenfalls ist unser aktuelles Wissen über ein gesundes Lungenmikrobiom limitiert. Unsere neueste Erkenntnis, dass das CF-Pathogen P.-aeruginosa auch in geringen Mengen im gesunden Lungenmikrobiom bei Kleinkindern auftauchen kann, ohne dass eine medizinische Intervention benötigt wird, zeigt, wie wichtig es ist, klinischrelevante Grenzwerte festzulegen. Solange diese Standardrichtlinien und Grenzwerte nicht vorhanden sind, bleibt die kultur-abhängige Diagnostik ein unersetzbares Verfahren, um das harmlose und nicht persistente Erscheinen geringer Mengen an Pathogenen von einer krankhaften Besiedlung der unteren Atemwege zu unterscheiden.
Literatur:
1 Charalampous T et al.: Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol 2019; 37: 783-92 2 Langdon A et al.: The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med 2016; 8(1): 39 3 Losada PM et al.: The cystic fibrosis lower airways microbial metagenome. ERJ 2019; 2(2): 00096-2015 4 Pienkowska K et al.: Airway microbial metagenomics. Microbes and Infection 2018; 20(9): 536-42 5 Bacci G et al.: A different microbiome gene repertoire in the airways of cystic fibrosis patients with severe lung disease. Int J Mol Sci 2017; 18(8): 1654 6 Pienkowska K et al.: Metagenome-inferred bacterial replication rates in cystic fibrosis airways. J Cyst Fibros 2019; 18(5): 653-56 7 Glassing A et al.: Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog 2016; 8: 24 8 Salter SJ et al.: Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 2014; 12: 87
Das könnte Sie auch interessieren:

Ausgewählte Mitteilungen und Poster
Am Jahreskongress der Schweizerischen Gesellschaft für Pneumologie vom 15. bis 16. Mai 2025 in Genf gaben Schweizer Pneumologinnen und Pneumologen einen Einblick in ihre vielfältige ...


Biologika in der Asthmatherapie
Was ist zu beachten bei der Biologikatherapie für Menschen mit Asthma? Wann sollte sie sinnvollerweise begonnen werden und wie lange sollte sie fortgesetzt werden? Prof. Dr. med. ...