IgA-Nephropathie: Update zu Pathogenese und Therapie
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die IgA-Nephropathie (IgAN) ist eine der häufigsten Ursachen primärer glomerulärer Erkrankungen weltweit. Die erfreulichen Fortschritte der letzten Jahre im Verständnis der Pathogenese ermöglichen einige neue therapeutische Ansätze. Im folgenden Artikel möchten wir deshalb die wichtigsten Fortschritte erläutern.
Keypoints
-
Die IgA-Nephropathie gehört weltweit zu den häufigsten glomerulären Erkrankungen und Ursachen des terminalen Nierenversagens.
-
Die Therapieentscheidung soll anhand einer Prognoseabschätzung erfolgen. Hierbei kann das «international prediction tool» Unterstützung bieten, der Nutzen ist jedoch noch nicht etabliert.
-
Maximale supportive Therapie bleibt Kern der Behandlung. Hierzu zählen RAAS-Blockade, optimale Blutdruckeinstellung, Gewichtsreduktion und Rauchstopp. Der Einsatz von SGLT2-Hemmern erscheint vielversprechend, hierzu sind weitere Daten zu erwarten.
-
Generell gilt es, Steroide und Immunsuppressiva zurückhaltender einzusetzen.
-
Gemäss neuen pathogenetischen Aspekten könnten Therapiestrategien wie Komplementsystem- und BAFF/APRIL-Inhibitoren eine vielversprechende Alternative zur bisherigen Immunsuppression bieten.
Klinik und Diagnose
Die geschätzte Inzidenz der IgAN liegt bei 2,5 pro 100000 Einwohner, wobei diese je nach geografischer Lage stark variiert; es zeigt sich eine niedrigere Prävalenz in Nordamerika und Europa und die höchste Prävalenz in Asien. In asiatischen Ländern sind Männer und Frauen gleichermassen betroffen, innerhalb von Europa und Nordamerika sind Männer häufiger betroffen als Frauen (3:1).1–3 Die Hälfte der Patienten verbleibt zunächst für einen längeren Zeitraum asymptomatisch. Die Erkrankung betrifft vorwiegend junge Erwachsene, kann aber grundsätzlich in allen Altersgruppen auftreten. Die Diagnose wird häufig erst gestellt, wenn die Nierenfunktion bereits eingeschränkt ist.
Zu den häufigsten klinischen Symptomen im Erwachsenenalter gehören die asymptomatische Hämaturie und die Proteinurie, die in 40–50% der Fälle im Anschluss an einen respiratorischen Infekt auftreten. Nur selten präsentieren sich die Betroffenen mit einem nephrotischen Syndrom oder einer rasch progredienten Glomerulonephritis. 25–30% der Erkrankten entwickeln innerhalb von 20–25 Jahren ein terminales Nierenversagen. Bei Auftreten von extrarenalen Symptomen, wie einer palpablen Purpura, Arthralgien und gastrointestinalen Beschwerden, spricht man von einer IgA-Vaskulitis.4,5
Für die Diagnosestellung einer IgAN ist eine Nierenbiospie zwingend erforderlich. Diese zeigt typischerweise mesangiale IgAAblagerungen. Lichtmikroskopisch zeigen sich unter anderem eine mesangiale Proliferation und endokapilläre Hyperzellularität, eine Tubulusatrophie und Fibrose sowie möglicherweise eine proliferative Glomerulonephritis mit Halbmonden. Die Immunfluoreszenz weist mesangiale Ablagerungen mit hauptsächlich IgA und vereinzelt IgG und IgM in sklerosierten Arealen auf. In 90% der Fälle ist ebenso C3 nachweisbar. Die histologische Klassifikation erfolgt nach dem Oxford-MEST-C-Score.6,7
Prognose
Der klinische Verlauf einer IgAN ist durchaus variabel, sodass Therapieentscheidungen idealerweise anhand einer Prognoseabschätzung und individuell getroffen werden sollten. Bisher sind keine Serum- oder Urinbiomarker hierfür vorhanden. Generell gilt eine Proteinurie von 1g/24Std. als einer der wichtigsten Risikofaktoren, da diese mit einem erhöhten Risiko für einen Nierenfunktionsverlust assoziiert ist. Weitere Risikofaktoren sind arterielle Hypertonie, Adipositas und Nikotinabusus. Die bisherige Risikostratifizierung erfolgte anhand des Ausmasses der Proteinurie, der Nierenfunktion und der bestehenden Risikofaktoren. Kürzlich wurde von Barbour et al. ein internationales «IgAN Prediction Tool» entwickelt, welches das Risiko für einen GFR-Abfall von mehr als 50% oder die Entwicklung eines terminalen Nierenversagens innerhalb von durchschnittlich 6,7 Jahren abzuschätzen versucht ( https://qxmd.com/calculate/calculator_499/international-igan-prediction-tool-adults# ).8 Hierbei werden klinische, laboranalytische und histopathologische Parameter miteinbezogen. Dieses Tool wurde allerdings nicht für den Gebrauch von Therapieentscheidungen validiert und ist nur zum Zeitpunkt der Nierenbiopsie verwendbar. Letztlich fehlen weitere randomisierte, kontrollierte Studien, um das «International IgAN Prediction Tool» als Grundlage für Therapieentscheidungen verwenden zu können. Allerdings stellt es einen wichtigen Ausgangspunkt für ein «shared decision making» und für die Erwägung von Vor- und Nachteilen bestimmter Therapiestrategien dar.8
Pathogenese
Multi-Hit-Modell
Es ist mittlerweile gut bekannt, dass IgAN-Patienten eine erhöhte Zirkulation von untergalaktosylierten IgA1 (Gd-IgA1) aufweisen, deren Quantität mit der Krankheitsschwere korreliert. Die in der «hinge region» untergalaktosylierten IgA1 werden normalerweise von mukosalen B-Zellen produziert. So wird vermutet, dass bei IgAN-Patienten nach einer mukosalen Infektion «primed» B-Zellen fälschlicherweise in die systemische Zirkulation gelangen und dort die «mukosalen» IgA1 produzieren. Die Gelenkregion («hinge region») fungiert aufgrund der freiliegenden N-Acetylgalactosamine (GalNAc) bei fehlender Galaktosebindung als Neoepitop und fördert die Bildung von Antiglykan-IgG-Autoantikörpern. Die Gd-IgA1 und die Autoantikörper bilden Immunkomplexe und aufgrund der Affinität der Gd-IgA1 zur extrazellulären Matrix führt dies zur Immunkomplexablagerung im Mesangium. Die Immunkomplex-Rezeptoren CD71 und der lösliche Rezeptor sCD89 können die Immunkomplexe innerhalb des glomerulären Mesangiums detektieren. Dies führt zur Freisetzung von proinflammatorischen, profibrotischen und proliferativen Mediatoren im Mesangium und somit zur Hyperproliferation der Mesangialzellen und Schädigung der Podozyten sowie Tubuluszellen in der Niere (Abb. 1).9,10
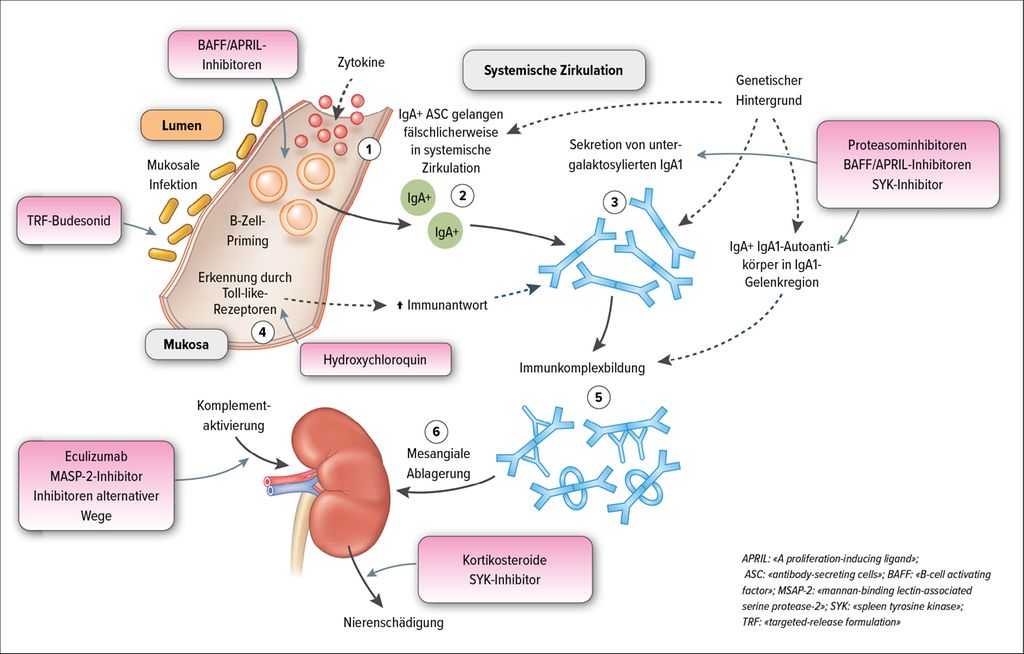
Abb. 1: Vorgeschlagene Pathogenese der IgA-Nephropathie und entsprechende Therapieansätze(modifiziert nach Floege et al.: Kidney Int 2019; 95: 268-80)
Neue Aspekte
In den letzten Jahren wurden einige neue Aspekte zur Pathogenese der IgAN geklärt. Die IgA1-Produktion von Plasmazellen wird durch T-Zell-Interaktionen und Zytokine hervorgerufen. Der zytokine «A proliferation inducing ligand» (APRIL) und der B-Zell-Aktivierungsfaktor aus der TNF-Gruppe (BAFF) führen zur Reifung von B-Zellen. Im Tierexperiment entwickelten Mäuse, die BAFF überexprimieren, eine IgAN mit einer erhöhten IgA-Serumkonzentration und mesangiale IgA-Ablagerungen bei gleichzeitig bestehender mikrobieller Besiedlung. Weiterhin konnte APRIL mit der Entstehung der IgAN in Verbindung gebracht werden, nicht zuletzt da APRIL und BAFF denselben Rezeptor verwenden. Diese Zytokine spielen also eine zentrale Rolle, um nach mikrobiellem Kontakt den Klassenwechsel von B-Zellen in IgA-produzierende Plasmazellen zu vollziehen. Im experimentellen Setting konnte eine Assoziation zwischen APRIL und der Produktion von IgA, gd-IgA und IgA-IgG-Immunkomplexen gezeigt und durch die Aktivierung von Toll-like-Rezeptor-9 (TLR-9) gesteigert werden. Letztendlich korrelierte die TLR-9-induzierte aberrante APRIL-Expression in den Tonsillen der IgAN-Patienten mit dem Ausmass der Proteinurie und der Kreatininerhöhung (Abb. 1).11,12
Die wachsende Datenlage bestätigt die Rolle des Komplementsystems in der Pathogenese der IgAN. Die GWAS («genome-wide association studies») haben einige genetische Risikoallele wie Komplementfaktor H (CFH) oder CFH-verwandte Proteine (CFHR1–5) identifiziert, die an der Regulation des alternativen Komplementwegs beteiligt sind. CFHR sind sequenziell ähnlich mit CFH und können mit diesen konkurrieren. So konnten Deletionen in den CFHR1- und CFHR3-Loci mit einem reduzierten Risiko für eine IgAN in Verbindung gebracht werden, eine erhöhte CFHR5-Plasmakonzentration scheint hingegen das Risiko zu erhöhen. Hinweise für die Beteiligung des Komplementsystems an der Pathogenese liefern ebenfalls die Nachweise von Bestandteilen der Komplementkaskade in den Nierenbiopsien. Komponenten des alternativen Komplementsystems wie C3 können in 90% der Biopsien im Mesangium gefunden werden. Des Weiteren war auch der Komplementfaktor H (CFH) in glomerulären Ablagerungen nachweisbar. Teilweise konnten auch Produkte des MBL(Mannose-bindendes Lektin)-Wegs, beispielsweise MBL-assoziierte Proteasen und C4d, dargestellt werden. MBL-Ablagerungen wurden grundsätzlich in Zusammenhang mit einer höheren Proteinurie und progredienten Nierenschädigung beobachtet (Abb. 2).3,13,14
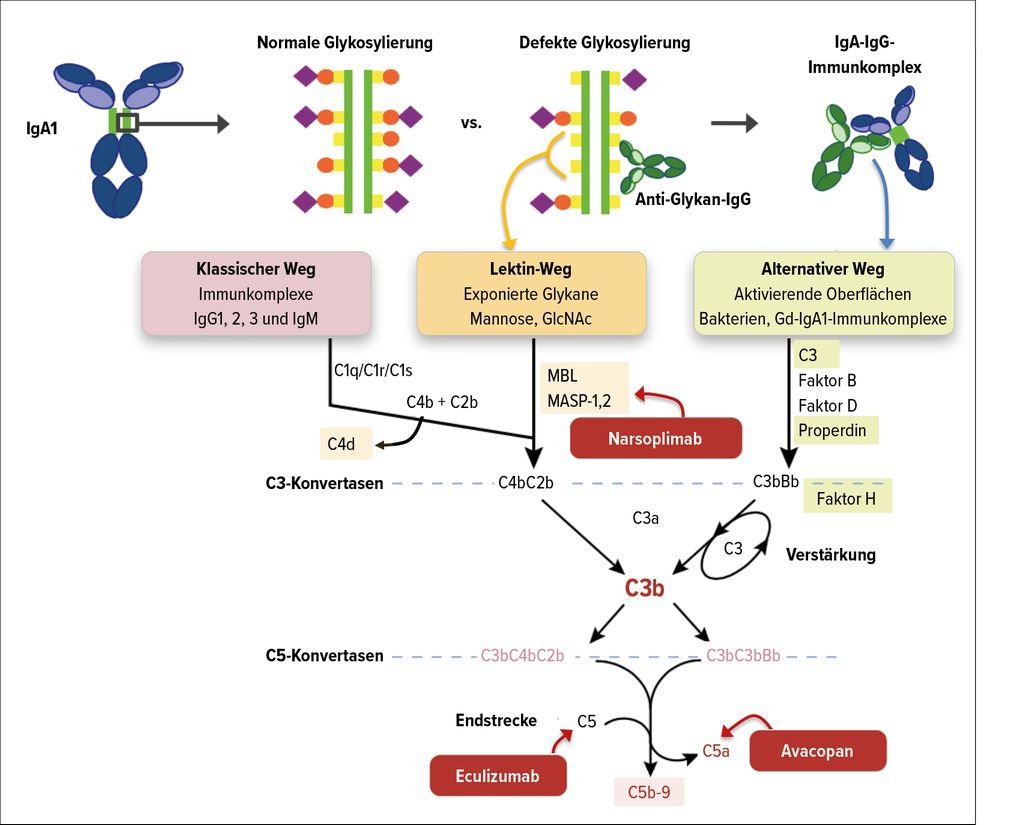
Abb. 2: Rolle des Komplementsystems in der Pathogenese der IgA-Nephropathie und potenzielle Therapieansätze (modifiziert nach: Rizk DV et al. und Thurman JM, Nester CM)13, 14
Therapie
Maximale supportive Therapie
Die IgAN ist in der Regel eine langsam fortschreitende Erkrankung, weshalb die Einstellung aller Risikofaktoren von grosser Bedeutung ist. Hierzu gehören eine optimale Einstellung des Blutdrucks, eine Gewichtsreduktion, genügend Bewegung sowie der Rauchstopp (Tab. 1). Einer der wichtigsten Bestandteile der supportiven Therapie ist die RAAS-Blockade zur Senkung der Proteinurie und Einstellung des Blutdrucks. Die KDIGO-Leitlinien von 2021 empfehlen die Etablierung einer RAAS-Blockade bei allen IgAN-Patienten mit einer Proteinurie >0,5g/d unabhängig davon, ob eine arterielle Hypertonie besteht. Bei persistierender Proteinurie von >1g/d unter maximal ausgebauter supportiver Therapie von 3 Monaten besteht ein erhöhtes Progressionsrisiko, sodass der Einsatz einer immunsuppressiven Therapie erwogen werden kann, insbesondere eine Steroidtherapie für die Dauer von 6 Monaten bei entsprechender eGFR und unter Einbezug der bestehenden Risikofaktoren (Diabetes mellitus, Osteoporose etc.).15

Tab. 1: Supportive Therapie der IgA-Nephropathie gemäss KDIGO-Leitlinie 202115
Systemische vs. lokale Glukokortikoide
Die STOP-IgAN-Studie untersuchte die Auswirkungen einer zusätzlichen immunsuppressiven Therapie zur supportiven Therapie hinsichtlich primärer Endpunkte, unter anderem bestehend aus Proteinurie und Nierenfunktionsverlauf. Hierbei handelte es sich um eine multizentrische, randomisierte, kontrollierte Studie, bei der 162 Patienten mit einer Proteinurie >0,75g/d nach 6-monatiger supportiver Therapie entweder mit einer Kombination aus supportiver und immunsupressiver Therapie oder mit einer rein supportiven Therapie behandelt wurden. 55 Patienten in der Immunsuppressionsgruppe hatten eine eGFR von >60ml/min/1,73m2 und erhielten eine Steroidmonotherapie, 27 hatten eine eGFR von 30–59ml/min/1,73m2 und erhielten zunächst für drei Monate eine Kombination aus Prednison und Cyclophosphamid, gefolgt von Azathioprin und Prednison bis zum Studienende nach 3 Jahren. Nach 3 Jahren zeigte sich bei den Patienten mit zusätzlicher Immunsuppression unabhängig von der GFR ein Benefit hinsichtlich der Reduktion der Proteinurie. Die Nierenfunktion war in beiden Gruppen jedoch nicht unterschiedlich, der GFR-Abfall von nur 4,2ml/min/1,73m2 in der Gruppe mit rein supportiver Therapie bestätigt den Nutzen der supportiven Therapie. Im Langzeit-Follow-up von zehn Jahren konnte kein zusätzlicher Benefit in der Immunsuppressionsgruppe aufgezeigt werden, hingegen eine erhöhte Anzahl von unerwünschten Ereignissen.16,17
Die TESTING-Studie untersuchte als multizentrische, doppelblinde, randomisierte, kontrollierte Studie Nutzen und Sicherheit der Steroidtherapie bei 262 Patienten mit IgAN. Hierbei wurden Patienten mit einer persistierenden Proteinurie >1g/d und einer eGFR von 20–120ml/min/1,73m2 nach 3-monatiger supportiver Therapie randomisiert und für 2 Monate mit Methylprednisolon 0,6–0,8mg/kg behandelt mit anschliessendem Ausschleichen über 4–6 Monate. Aufgrund einer Vielzahl von unerwünschten Ereignissen, vor allem Infektionen, musste die Studie vorzeitig abgebrochen werden. Zu diesem Zeitpunkt fiel jedoch der primäre Endpunkt (Reduktion der eGFR um 40%, Nierenversagen, renaler Tod) zugunsten der Steroidtherapie aus.18
Die randomisierte, kontrollierte NEFIGAN-Studie untersuchte die Wirkung von TRF(«targeted-release formulation»)-Budesonid als topisch wirksame Therapie vs. Placebo. Die Freisetzung im distalen Ileum soll die lokale Produktion von Gd-IgA1 reduzieren. Eingeschlossen wurden Patienten mit einer eGFR >45ml/min/1,73m2 und einer persistierenden Proteinurie von >0,75g/d trotz durchgeführter 6-monatiger RAAS-Blockade. In der Behandlungsgruppe zeigte sich eine Reduktion der Proteinurie um 24,4% bei stabiler eGFR, jedoch traten auch systemische Nebenwirkungen wie cushingoide Merkmale, Akne und Schlafstörungen auf, die insgesamt zu einer Drop-out-Rate von 12% führten.19Die Ergebnisse der laufenden Phase-III-Studie NefIgArd sind noch ausstehend.
Andere Immunsuppressiva
Alternative immunsuppressive Substanzen wie Azathioprin und Rituximab konnten in bisherigen Studien keinen signifikanten Benefit zeigen und werden demnach nicht empfohlen.20,21 Ein Nutzen von Mycophenolat mofetil oder Hydroxychloroquin konnte hauptsächlich bei asiatischen Patienten nachgewiesen werden, sodass diese Therapien bei anderen Ethnizitäten nicht klar empfohlen werden können.22–26
Neue Therapien
Im Hinblick auf die neuen Erkenntnisse in der Pathogenese untersucht eine Vielzahl von Studien neue Therapieansätze, sodass in den nächsten Jahren spannende Ergebnisse erwartet werden können.
Die neuen Erkenntnisse hinsichtlich der Beteiligung der Komplementkaskade in der Pathogenese liefern einige Ansätze für neue Therapiestrategien. Hierzu laufen aktuell klinische Studien zum Einsatz neuer Medikamente, die den Lektin-Weg (MASP-2-Inhibitor Narsoplimab), den alternativen Weg (Faktor-B-Inhibitor LNP023) und die gemeinsame Endstrecke (C5-Komplementinhibitor Cemdisiran und C5a-Antagonist Avacopan) des Komplementsystems betreffen, deren Ergebnisse noch ausstehend sind.27 Weitere klinische Studien untersuchen Therapieformen bezüglich der B-Zell-Reifung unter Einbezug von BAFF und APRIL und beinhalten den BAFF-Antagonisten Blisibimod (BRIGHT-SC trial) und das TACI-Ig-Fusionprotein Atacicept. Ein weiterer Ansatz ist die kombinierte Angiotensin- und Endothelin-Rezeptorblockade mittels Sparsentan (Abb. 1 und 2).3
Vielversprechend ist ebenfalls der Einsatz von SGLT2-Hemmern bei IgAN-Patienten ohne Diabetes. In der DAPA-CKD-Studie konnte eine signifikante Reduktion des primären Endpunkts, bestehend aus 50% Abfall der eGFR, Nierenversagen und Tod, unter 10mg Dapagliflozin im Vergleich zu Placebo gezeigt werden.28,29 Hierbei handelte es sich um eine vordefinierte Analyse der DAPA-CKD-Studie von 270 der insgesamt 4094 Patienten, die eine IgAN mit einer mittleren eGFR von 43,8±12,2ml/min/1,73m2 und eine Albuminurie von 540–1515mg/g aufwiesen. 3,6% Patienten in der DAPA-Gruppe erreichten den renalen Endpunkt vs. 15% in der Placebogruppe. Bei SGLT2-Hemmern geht man von zahlreichen Wirkmechanismen aus. Hierbei ist die Wiederherstellung des tubuloglomerulären Feedbacks, die eine geringere Hyperfiltration zufolge hat und eine metabolische Stressreduktion der proximalen Tubuluszellen durch weniger Rückresorption bewirkt, eines von vielen nennenswerten Wirkprinzipien.30 Die weiteren Ergebnisse der laufenden Studien, z.B. EMPA-KIDNEY, werden mit Spannung erwartet.
Literatur:
1 Wyatt RJ, Julian BA: N Engl J Med 2013; 368: 2402-14 2 Magistroni R et al.: Kidney Int 2015; 88: 974-89 3 Seikrit C et al.: Nephrol Dial Transplant 2021; 36(Suppl2): 24-30 4 Szeto CC et al.: Am J Med 2001; 110: 434-7 5 Audemard-Verger A et al.: Arthritis Rheumatol 2017; 69: 1862-70 6 Pattrapornpisut P et al.: Am J Kidney Dis 2021; 78: 429-41 7 Trimarchi H et al.: Kidney Int 2017; 91: 1014-21 8 Barbour SJ et al.: JAMA Intern Med 2019; 179: 942-52 9 Boyd JK et al.: Kidney Int 2012; 81: 833-43 10 Suzuki H et al.: J Am Soc Nephrol 2011; 22: 1795-803 11 McCarthy DD et al.: J Clin Invest 2011; 121: 3991-4002 12 Muto M et al.: J Am Soc Nephrol 2017; 28: 1227-38 13 Rizk DV et al.: Front Immunol 2019; 10: 504 14 Thurman JM, Nester CM: Clin J Am Soc Nephrol 2016; 11: 1856-66 15 Kidney Disease: Improving Global Outcomes Glomerular Diseases Working Group:Kidney Int 2021; 100(4S): S1-S276 16 Rauen T et al.: N Engl J Med 2015; 373: 2225-36 17 Rauen T et al.: Kidney Int 2020; 98: 1044-52 18 Lv J et al.: JAMA 2017; 318: 432-42 19 Fellström BC et al.: Lancet 2017; 389: 2117-27 20 Pozzi C et al.: J Am Soc Nephrol 2010; 21: 1783-90 21 Lafayette RA et al.: J Am Soc Nephrol 2017; 28: 1306-13 22 Tang S et al.: Kidney Int 2005 68: 802-12 23 Frisch G et al.: Nephrol Dial Transplant 2005; 20: 2139-45 24 Tang SC et al.: Kidney Int 2010; 77: 543-549 25 Du B et al.: BMC Nephrol 2017; 18: 245 26 Liu LJ et al.: Am J Kidney Dis 2019; 74: 15-22 27 Lafayette RA et al.: Kidney Int Rep 2020; 5: 2032-41 28 Wheeler DC et al.: Kidney Int 2021; 100: 215-24 29 Heerspink HJ et al.: N Engl J Med 2020; 383: 1436-46 30 Anders HJ et al.: Nephrol Dial Transplant 2020; gfaa329
Das könnte Sie auch interessieren:
Neue Klassifikation soll für mehr Klarheit sorgen
Die Glomerulonephritis ist eine komplizierte Angelegenheit. Das liegt auch daran, dass die immunvermittelten Erkrankungen anhand von histopathologischen Mustern beschrieben werden, die ...

Therapie der ANCA-assoziierten Vaskulitis: Gibt es steroidfreie Alternativen?
Noch in den 1950er-Jahren verstarben rund 90% der Patient:innen, die an einer mit antineutrophilen zytoplasmatischen Antikörpern (ANCA) assoziierten Vaskulitis litten, im ersten Jahr ...
Spannende Fälle
Neben ihren Forschungsergebnissen stellten Schweizer Nephrologinnen und Nephrologen am Jahreskongress 2024 in Basel auch einige spannende und lehrreiche Fälle vor. Wir präsentieren Ihnen ...