
Was ist bei neonatalen Anfällen zu tun?
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Bislang fehlt eine einheitliche Leitlinie zur Versorgung epileptischer Anfälle bei Neugeborenen. Die hier präsentierte Richtlinie soll Klarheit in Bezug auf die Klassifikation, Ätiologie, Diagnostik und Therapie dieser Anfälle schaffen.
Keypoints
-
Ein MRI des Schädels weist in 70% der neonatalen Anfälle die Ursache aus.
-
Die häufigsten Ursachen sind die hypoxisch-ischämische Enzephalopathie (30–50%) und zerebrale Blutungen (20–25%).
-
Sowohl klinische wie elektroenzephalografische Anfälle müssen behandelt werden.
-
Die Anfallssemiologie kann hinweisend auf die Ätiologie sein und sollte bei der Medikamentenwahl berücksichtigt werden.
Die Häufigkeit neonataler Anfälle ist in im Vergleich zu jedem weiteren Lebensabschnitt am höchsten. Sie sind meist symptomatisch, wobei die hypoxisch-ischämische Enzephalopathie (30−50%) und zerebrale Blutungen (20−25%) am häufigsten auslösend sind. Alle anderen Ursachen sind demgegenüber selten, haben jedoch wegen möglicher therapeutischer Konsequenzen besondere Bedeutung. Dazu zählen unter anderem Hirnfehlbildungen, genetische Epilepsien, Infektionen, Hypoglykämien, Elektrolytentgleisungen und Stoffwechselstörungen.
Die noch zu publizierende Richtlinie für neonatale Anfälle aus dem Inselspital Bern gliedert sich in die Bereiche Klassifikation, Ätiologie, Diagnostik und Therapie. Die wichtigsten Aspekte aus diesen Teilgebieten werden im Folgenden präsentiert.
Klassifikation1
Bei neonatalen Anfällen gilt in erster Linie zu bedenken, dass sie immer fokal sind, weil die axonale Propagation noch nicht vorhanden ist.
Elektroklinisch ist eine Vielzahl an Anfallstypen zu beobachten. Zu den motorischen Symptomen zählt etwa das Auftreten von Automatismen. Klonische Anfälle sind typisch bei einem Schlaganfall bzw. einer Hirnblutung und auch bei hypoxisch-ischämischer Enzephalopathie (HIE) möglich. Epileptische Spasmen sind sehr selten zu beobachten und geben Anlass, an Pyridoxin-abhängige oder andere metabolische Epilepsien zu denken. Myoklonische Anfälle treten bei Frühgeborenen oder Neugeborenen mit Stoffwechseldefekten auf. Tonische Anfälle (fokal, unilateral oder bilateral asymmetrisch) sind typisch bei EIEE («early-infantile epileptic encephalopathy») und genetischen Neugeborenenepilepsien. Generalisierte Tonisierung (z.B. Opisthotonus) ist hingegen nicht epileptisch.
Nichtmotorische Anfälle treten selten isoliert auf, ein aEEG bzw. EEG ist hier obligat. Sie können sich autonom bemerkbar machen, also das vegetative Nervensystem betreffen, oder als Verhaltensarrest.
Sequenzielle Anfälle mit typischer Abfolge (die Seite kann allerdings wechseln) sind oft bei genetischen oder metabolischen Epilepsien, z.B. Ionenkanalerkrankungen (KCNQ2/3, SCN2A), zu beobachten.
Anfälle, die nur elektrografisch festzustellen sind, finden sich oft bei Frühgeburten oder HIE (v.a. bei Läsionen in den Basalganglien/im Thalamus), kritisch kranken Neugeborenen und Neugeborenen mit kardiochirurgischem Eingriff.
Elektroenzephalografische Kriterien für einen Neugeborenenanfall sind:2
-
plötzliche Veränderung im EEG
-
repetitive Wellenformen bzw. Potenziale, die sich bezüglich Morphologie, Frequenz und/oder Lokalisation entwickeln und sich von der übrigen EEG-Aktivität abgrenzen
-
Amplitude: mind. 2µV
-
Dauer: mind. 10 Sekunden (Anmerkung: Diese Dauer ist willkürlich gewählt)
-
Korrelation mit klinischen Anfallskriterien (kann, muss aber nicht vorliegen)
Ätiologie3–5
Wegen der unmittelbaren therapeutischen Konsequenzen sind stets eine Bestimmung von Glukose (und falls kleiner als 2,5mmol/L, zusätzlich «critical sample») und ionisiertem Ca2+ im Blut indiziert. Dies gilt auch, wenn neonatale Anfälle im Rahmen einer HIE nach perinataler Asphyxie (30–50%) auftreten.6 In 25–30% der Fälle sind zerebrovaskuläre Ursachen verantwortlich, die zu Anfällen führen, die bei reifen Neugeborenen in den ersten 1–3 Lebenstagen zum Teil ohne zusätzliche neurologische Begleitsymptomatik im Intervall zu beobachten sind. Sie sind vor allem verdächtig für arterielle ischämische Infarkte, eine Subarachnoidalblutung oder eine Sinusvenenthrombose. Eine umgehend durchgeführte Ultraschalluntersuchung ist sinnvoll, aber nicht ausreichend sensitiv. Ein MRT des Schädels mit diffusionsgewichteten Sequenzen (ohne Kontrastmittel) ist in 70% der Fälle diagnostisch zielführend. Hirnfehlbildungen sind für ca. 5% der Fälle verantwortlich.4
Anfälle, die in den ersten Lebenstagen zusammen mit einer progredienten neurologischen Symptomatik auftreten, deuten auf eine angeborene Stoffwechselstörung hin.4 Beispiele sind etwa:
-
Störungen im Pyridoxin-Stoffwechsel: Sie führen zu heftigen, mit anfallssupprimierenden Medikamenten schlecht beherrschbaren Anfällen. Klinisch treten Spasmen bzw. Anfälle mit tonischer Komponente und fazialer Betonung auf. Zu den häufigsten zugrunde liegenden Störungen zählen Mutationen im ALDH7A1-Gen, das für das sogenannte Antiquitin (Aminoadipinsemialdehyd, AASA) kodiert. Seltenere Ursachen sind Defekte der Pyridoxamin-5-Phosphat-Oxidase oder eine Hypophosphatasie, welche die Verfügbarkeit des aktiven Metaboliten Pyridoxal-Phosphat herabsetzen. Diagnostisch wegweisend ist die Bestimmung der Pipecolsäure-Konzentration im Plasma, AASA im Urin und der alkalischen Phosphataseaktivität im Serum (stark erniedrigt bei Hypophosphatasie). Zudem kommt es bei ALDH7A1-Defekten gehäuft zu perinatalen Störungen, die mit den auftretenden Anfällen in Zusammenhang gebracht werden, etwa Hypokalzämie, Hypomagnesiämie, Diabetes insipidus, Hypothyreose oder niedrige Apgar-Werte. ALDH7A1-Defekte liegen auch dem klinischen Krankheitsbild der Folinsäure-responsiven Anfälle zugrunde.7
-
Biotin-Mangel: verursacht durch Holocarboxylase-Synthetase- oder Biotinidase-Defekte, diese sprechen auf orales Biotin an.7 Der Biotinidase-Defekt ist in der Schweiz im Neugeborenenscreening eingeschlossen.
-
Molybdän-Kofaktor-Mangel: Dieser geht mit erhöhten Sulfiten im Urin einher. Typ A spricht auf intravenöses cPMP (zyklisches Pyranopterin-Monophosphat) an.
-
DEND (developmental delay, epilepsy and neonatal diabetes): Dieses Syndrom wird nicht mit Insulin, sondern mit hypoglykämischer Nahrung/Sulfonylurea behandelt.8,9
Neonatale Anfälle sind in 15–20% der Fälle auf Infektionen zurückzuführen. In der gesamten Neonatalperiode können Anfälle Ausdruck einer Meningitis sein. Hauptverantwortlich sind hierfür B-Streptokokken und gramnegative Enterobacteriae. Ab der zweiten Lebenswoche − sehr selten früher − kann auch eine virale Enzephalitis durch das Herpes-simplex-Virus oder Enteroviren ursächlich sein. Beweisend ist nur der Erregernachweis mittels Kultur oder PCR im Liquor. Glukose-, Zellzahl- und Eiweissbestimmungen im Liquor sind bei der neonatalen Meningitis unzuverlässig.4
Hypomagnesiämien mit heftigen Krampfanfällen treten ab der zweiten bis dritten Lebenswoche infolge hereditärer Magnesium-Transport-Defekte (z.B. im TRPM6-Gen) oder nach intestinalen Verlusten (Stomaanlage) auf. Meist besteht zeitgleich eine mässige Hypokalzämie, die Kalziumsubstitution alleine vermag die Anfälle aber nicht nachhaltig zu unterbrechen.4
In 4% der Fälle sind Drogen- bzw. Medikamentenmissbrauch für neonatale Anfälle ursächlich. Nach mütterlichem Opiatkonsum in der Schwangerschaft kann es als Ausdruck eines neonatalen Drogenentzugs zu Anfällen kommen, die gut auf Morphin, nicht aber auf Phenobarbital ansprechen. Neonatale Anfälle sind auch nach mütterlicher Einnahme von selektiven Serotonin-Wiederaufnahme-Inhibitoren (SSRI), speziell Venlafaxin, Barbituraten oder Benzodiazepinen während der Schwangerschaft beschrieben.4,9
Genetische Epilepsiesyndrome, insbesondere Ionenkanalerkrankungen, machen 3–15% der Anfälle bei Neugeborenen aus. Sie treten bei Auffälligkeiten in Genen, die für zerebral exprimierte Kalium- oder Natriumionenkanäle kodieren (z.B. KCNQ2, KCNQ3, SCN2A), auf. Das klinische Spektrum ist sehr breit und reicht von selbstlimitierten Anfällen (neonatale Anfälle des 5. Lebenstages, familiäre Neugeborenenanfälle) bis zu neonatalen Enzephalopathien. Letztere zeigen sich als Epilepsie des Säuglingsalters mit wandernden fokalen Anfällen oder frühinfantile epileptische Enzephalopathie mit Burst-Suppression-Muster (Ohtahara-Syndrom). Eine Therapie mit hoch dosierten Natriumkanalblockern (Phenytoin) sollte bei schweren Formen mit Burst-Suppression-Muster früh versucht werden. (Cave: Verschlechterungen sind möglich, wobei Epilepsiesyndrome wie das Dravet-Syndrom bei SCN1A-Mutation, bei denen diese Medikation kontraindiziert ist, erst im Säuglingsalter manifest werden). Bei diesen Formen ermöglicht die genetische Diagnostik eine gezielte Therapie. Unauffälligkeit in der Anamnese oder im neurologischen Befund und interiktale Grundaktivität im EEG (epilepsietypische Signale sind möglich) sind suggestiv für selbstlimitierte Epilepsiesyndrome.
Veränderungen im SLC2A1-Gen, das für den Glukose-Transporter Glut1 kodiert, können am Ende der Neugeborenenperiode oder später zu zerebralen Anfällen führen, die auf Medikamente schlecht ansprechen. Diagnostisch wegweisend sind niedrige Glukosekonzentrationen im Liquor (<45% der unmittelbar vor Lumbalpunktion im Nüchternzustand gemessenen Blutglukosekonzentration). Die Therapie besteht in einer ketogenen Diät.4
Diagnostik & Check-up
An erster Stelle steht die sofortige Blutgasanalyse mit Bestimmung von Blutzucker und Ca2+.
In der klinischen Untersuchung wird Hinweisen auf eine Infektion oder syndromale Erkrankung nachgegangen. Eine Sonografie des Schädels mit Gefässdarstellung der Sinusvenen ist ebenfalls indiziert. Ein MRT des Schädels weist in 70% der Fälle die Ursache aus und hat mit Diffusion, Spektroskopie und Gefässdarstellung zu erfolgen.
Ein Standard-EEG inkl. eines bilateralen Deltoideus-EMG zur Erfassung von Myoklonien ist durchzuführen. Es besteht eine grosszügige Indikation für aEEG und Langzeit-Video-EEG, da subklinische Anfälle häufig sind. Die American Clinical Neurophysiology Society empfiehlt ein 24-Stunden-Video-EEG bei Hochrisikokindern bis zur 24-stündigen Anfallsfreiheit, zumal die Gabe von anfallssupprimierender Medikation zu einer klinisch-elektrophysiologischen Dissoziation mit nur subklinischen Anfällen führen kann. Das Anfallsrisiko bei HIE und Hypothermie beträgt 50%. Während der Kühlung und des Aufwärmens ist ein aEEG bis zur 24-stündigen Anfallsfreiheit obligat und, falls möglich, ein Langzeit-Video-EEG wünschenswert.6 Der akkurateste prädiktive Faktor für Anfälle ist eine schwere Hintergrundaktivität am Anfang der Ableitung.
Bei der Blutentnahme sind folgende Parameter zu bestimmen: Glukose, K, Na, Cl, Ca, Mg, Phosphat, BGA, Laktat (ggf. Pyruvat), Ammoniak, ASAT, ALAT, AP (cave: erniedrigt hinweisend auf kongenitale Hypophosphatasie), Kreatinin, Harnsäure, CRP, Differenzialblutbild und Blutkultur. Besteht weiterhin Unklarheit, bieten sich folgende zusätzliche Untersuchungen an:
Aminosäuren, Acylcarnitinprofil, Pipecolsäure, Homocystein, CDG, VLCFAs sowie HHV6- und TORCH-Serologie.
Zudem ist eine Urinkultur durchzuführen und es sind organische Säuren, AASA, Aminosäuren (mit Sulfocystein), Sulfit, Guanidinoacetat und Kreatin im Urin zu bestimmen; gegebenenfalls auch eine Toxikologie (bei Verdacht auf Entzug).
Die zu erhebenden Liquoruntersuchungen beinhalten: Kultur, PCR (HSV, Enteroviren), Zellzahl, Eiweiss, Glukose (mit gleichzeitiger (!) Ratio Liquor/Serum), Laktat, Aminosäuren, Neurotransmitter (cave: Liquormenge, Prioritätenliste). Cave: Ein Glut1-Defekt ist nur ausgeschlossen, wenn die Werte >4 Stunden postprandial, oder nach unveränderter intravenöser Glukosezufuhr, erhoben werden.8 Dieser wird allerdings in der Neonatalperiode nur sehr selten symptomatisch.
Die ophthalmologische Untersuchung auf Kolobom, Katarakt, Retinopathie oder Optikushypoplasie ist ebenfalls indiziert.
Schliesslich kann auch eine genetische Untersuchung Klarheit schaffen.
Bei Vitamin-B6-abhängigen Epilepsien kommt es zu spezifischen Laborauffälligkeiten.7 Bei Antiquitinmangel sinken die AASA-Werte im Urin unter Vitamin-B6-Gabe, bleiben jedoch im pathologischen Bereich. Da AASA auch bei Patienten mit Molybdän-Kofaktormangel erhöht ist,7 muss simultan zur Bestimmung der AASA auch jene von Sulfocystein erfolgen. Eine Erhöhung der Pipecolsäure im Plasma oder Liquor ist ebenfalls zuverlässig, allerdings weniger spezifisch, da die Pipecolsäure auch bei peroxisomalen Erkrankungen erhöht sein kann.7 Nach positivem Ansprechen auf Vitamin B6 bzw. Folsäure und bei Nachweis von erhöhtem AASA erfolgt die molekulare Untersuchung des Antiquitingens.5,11 Eine PNPO-Mutationsanalyse wird bei Patienten entweder mit Pyridoxin- oder PLP-responsiven Anfällen empfohlen, die normale AASA-Werte aufweisen.9
Therapie
Es gibt keine evidenzbasierten oder allgemein akzeptierten Leitlinien. Die Entscheidungen basieren auf klinischer Erfahrung, Beobachtungsstudien und einer begrenzten Anzahl randomisierter Studien.
Im Allgemeinen sind im Notfall die bekannten ABC-Massnahmen anzuwenden: Atemwege frei machen und offen halten («airway»), beatmen («breathing») und Herzdruckmassage («circulation»). Zusätzliche Massnahmen beinhalten das kardiorespiratorische Monitoring, die sofortige Korrektur transienter Elektrolytentgleisungen/Hypoglykämien und das Sicherstellen einer adäquaten Flüssigkeitssubstitution. Bei Verdacht auf eine Stoffwechselstörung sind eine Nahrungskarenz und Glukose 10% i.v. indiziert. Antibiotische und/oder antivirale Therapien sind bei Verdacht auf eine Infektion einzuleiten.
Anfallssupprimierende Medikation (ASM)
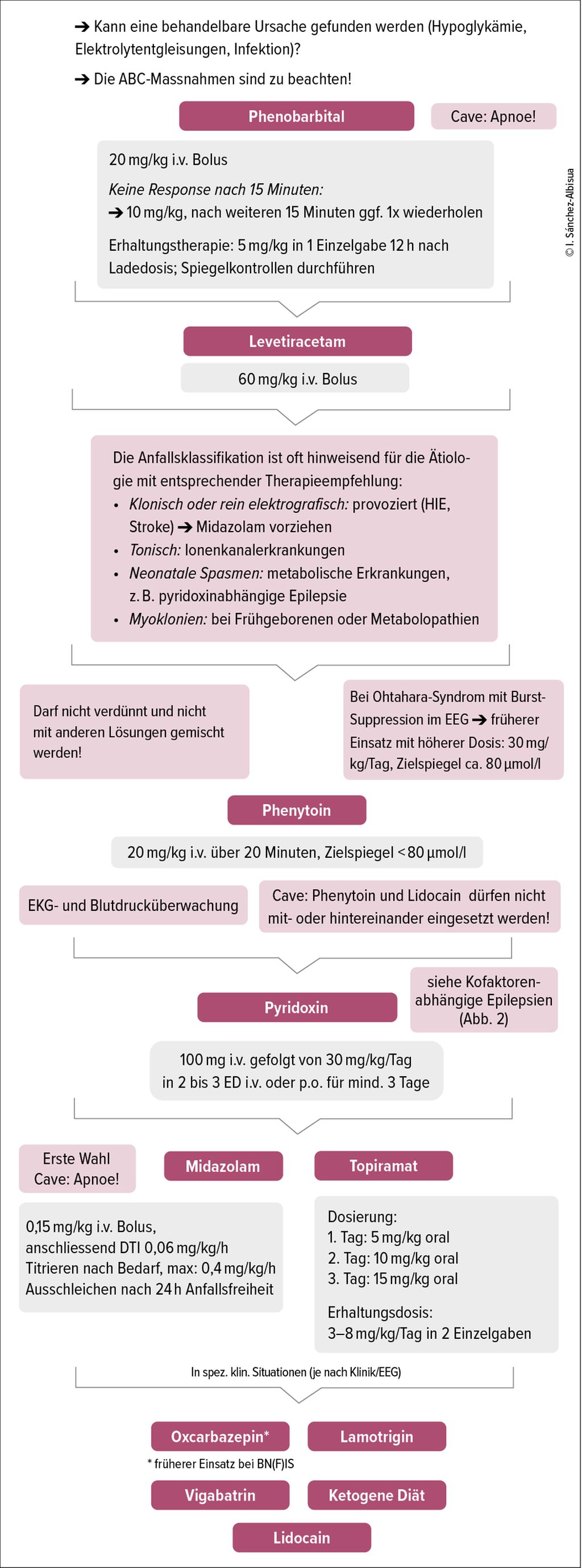
Abb. 1: Therapieschema für neonatale Anfälle
Sowohl klinische wie elektroenzephalografische Anfälle müssen behandelt werden. Die Anfallssemiologie kann hinweisend auf die Ätiologie sein und sollte bei der Medikamentenwahl berücksichtigt werden. Am Anfang ist eine Ladedosis bis zur Anfallsunterbrechung gemäss Fliessschema (Abb.1) zu wählen. Bei fehlender Wirksamkeit ist die Ätiologie zu reevaluieren. Zum Beispiel sollten Anfälle nach Asphyxie nach ein paar Tagen sistieren. Tritt das nicht ein, ist an genetische bzw. metabolische Ursachen zu denken.
Da sich die Pharmakokinetik bei Neugeborenen anders verhält als bei älteren Kindern, ist es sinnvoll, die Spitalpharmazie zu involvieren, insbesondere bei Polytherapie. Symptomatische Anfälle sistieren innert weniger Tage und rezidivieren selten, sodass eingeleitete Therapien rechtzeitig abzusetzen sind.
Medikamentöse Intervention6,10
Wir empfehlen den Therapie-Algorithmus gemäss Abbildung 1. Folgende Hinweise zur medikamentösen Therapie sind besonders zu beachten:
-
Phenobarbital ist wirksamer, aber toxischer als Levetiracetam.11 Angesichts der Nebenwirkungen stellt Levetiracetam bei Neonaten mit kardialer und/oder Leberdysfunktion eine gute Option dar.
-
Bei Ionenkanalerkrankungen mit Ohtahara-Syndrom ist ein Therapieversuch mit ausreichend dosiertem Phenytoin empfehlenswert (30mg/kg/Tag, Zielspiegel ca. 80µmol/l).
-
Bei der Verabreichung von Midazolam muss, abgesehen von der Sedation, auf Grund potentiell atemdepressiver und kardiovaskulärer Effekte die Möglichkeit zur intensivmedizinischen Überwachung inkl. Intubation gegeben sein.
-
Topiramat ist in ca. 50% der Fälle wirksam und wird gut vertragen.12 Eine i.v. zu verabreichende Lösung ist nicht verfügbar.
Therapie der Vitamin-B6-abhängigen Epilepsien7,10
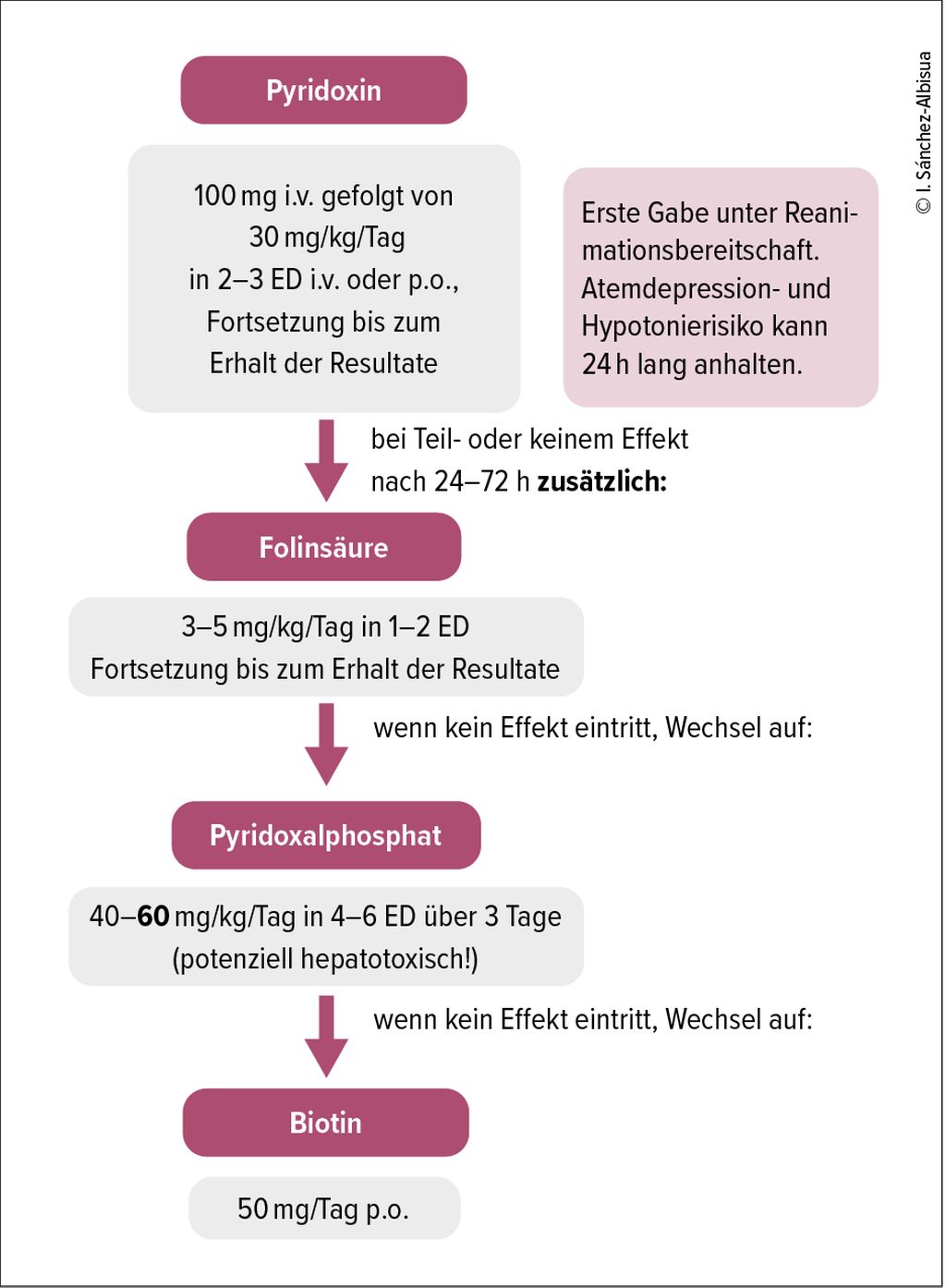
Abb. 2: Therapie Kofaktoren-abhängiger Epilepsien
Bei ätiologisch unklaren, therapieresistenten Anfällen sollte vom Neugeborenen bis zum frühen Kleinkindalter ein standardisierter Vitamin-B6-Versuch erfolgen. Der Antiquitinmangel spricht auf Pyridoxin an, während der viel seltenere Pyridox(am)in-5•-Phosphat-Oxidase(PNPO)-Mangel auf Pyridoxalphosphat anspricht (und in manchen Fällen bei Restaktivität auch auf Pyridoxin). In Anbetracht der grösseren Häufigkeit des Antiquitinmangels im Vergleich zum PNPO-Mangel sowie von Berichten über die potenzielle Lebertoxizität von Pyridoxalphosphat sollte für diesen standardisierten Vitamin-B6-Versuch in erster Linie Pyridoxin zur Anwendung kommen. Pyridoxalphosphat sollte aufgrund jüngster Berichte über Hepatopathien nur bei Pyridoxin-Non-Respondern eingesetzt werden.
Ein Pyridoxintest sollte beim Neugeborenen mit einer Dosis von 30mg/kg/Tag in 2–3 Einzeldosen über 3 aufeinanderfolgende Tage i.v. oder fakultativ p.o. durchgeführt werden. Die erste Gabe sollte aufgrund möglicher Apnoen unter Reanimationsbereitschaft erfolgen. Die klinische Response erfolgt innert Minuten, das interiktale EEG normalisiert sich nach 24–48h. Bei fehlender Wirksamkeit sollte individuell nach 24–72h eine Zusatztherapie mit Folinsäure, 3–5mg/kg/Tag als Einzeldosis, oder ein Wechsel zu Pyridoxalphosphat 40–60mg/kg/Tag in 4–6 Einzeldosen erwogen werden. PNPO-Patienten, welche unter Pyridoxin anfallsfrei sind, sollten nicht auf Pyridoxalphosphat umgestellt werden. Bei mangelnder Anfallsfreiheit unter Pyridoxin und gesichertem PNPO-Mangel sollte ein langsames, schrittweises Umstellen auf Pyridoxalphosphat über 3–4 Wochen erfolgen.
Die folinsäureabhängige Epilepsie ist genetisch mit dem Antiquitinmangel allelisch identisch. Die Folinsäure könnte evtl. einen «Einsparungseffekt» auf das Vitamin B6 haben. Daher kann der zusätzliche Einsatz von Folinsäure (3–5mg/kg/Tag) als Einzeldosis gerade bei Neugeborenen mit inkomplettem Ansprechen auf Pyridoxin sinnvoll sein.7
Biotinidase-Mangel
Ein Holocarboxylase-Synthetase-Defekt sollte mittels des typischen biochemischen Profils diagnostizierbar sein und mittels Enzymatik und Genetik bestätigt werden. Dazu müssen vor der ersten Gabe die entsprechenden Analysen abgenommen werden: BGA, Laktat, NH3, Aminosäuren im Plasma und organische Säuren im Urin. Die empfohlene Dosis beträgt 50mg/Tag,13 denn ein Biotinidase-Mangel, bei dem eine Dosis von 10mg/Tag genügt, wird bereits im Neugeborenen-Screening gesucht. Dieser würde zudem frühestens im Verlauf der ersten paar Monate zu Symptomen führen und bis zu diesem Zeitpunkt aber bereits behandelt sein. Alternativ könnte man auch mit Biotin 10mg/Tag starten und dann, bei Hinweisen in den Laboranalysen, aber ungenügendem Ansprechen, die Dosis weiter eskalieren.
Therapiedauer
Bei Neonaten mit akut provozierten Anfällen können die ASM nach wenigen Tagen Anfallsfreiheit abgesetzt werden. Eine längere Therapie verbessert die Prognose nicht.14 Bei Neonaten mit Epilepsiesyndromen sollten die ASM länger beibehalten werden.
Fazit
Epileptische Anfälle in der Neonatalperiode können neben der hypoxisch-ischämischen Enzephalopathie und zerebralen Blutungen zahlreiche weitere Ursachen haben. Dementsprechend ist eine Vielzahl an Anfallstypen zu beobachten. Eine sofortige Diagnostik ist essentiell, um primär reversible Ursachen wie Hypoglykämien, Elektrolytstörungen oder Infektionen unmittelbar behandeln zu können. Die Diagnostik umfasst neben standardmässigen EEG-Ableitungen auch eine MRT-Untersuchung bis hin zu Stoffwechseldiagnostik oder genetischen Untersuchungen in der erweiterten Abklärung. Das vorgeschlagene Therapieschema umfasst zunächst eine stufenweise Eskalation der anfallssupprimierenden Medikation. Die Anfallssemiologie kann jedoch hinweisend auf die Ätiologie sein und sollte daher bei der Medikamentenwahl berücksichtigt werden.
Literatur:
1 Pressler RM et al.: The ILAE classification of seizures and the epilepsies: Modification for seizures of the neonate. Position paper by the ILAE Task Force on Neonatal Seizures. Epilepsia 2021; 62: 615-8 2 Volpe J: Neonatal seizures, in: Volpe’s Neurology of the Newborn. 6. Auflage. Amsterdam: Elsevier, 2018 3 Panzer A et al.: Epilepsien bei Kindern und Jugendlichen. Bern: Huber, 2015 4 AWMF-Leitlinie: Zerebrale Anfälle bei Neugeborenen, RN 024-011 (angemeldetes Leitlinienvorhaben, geplante Fertigstellung: 31.03.2024) 5 Shellhaas R: Clinical features, evaluation, and diagnosis of neonatal seizures. www.uptodate.com (abgerufen im September 2020) 6 Shellhaas R: Treatment of neonatal seizures. www.uptodate.com (abgerufen im Juni 2020) 7 Plecko B, Abela L: Vitamin B6-abhängige Epilepsien – ein Update. Epileptologie 2016; 33: 102-9 8 Pearl PL: New treatment paradigms in neonatal metabolic epilepsies. J Inher Metab Dis 2009; 32: 204-13 9 Shellhaas R: Neonatal epilepsy syndromes. www.uptodate.com (abgerufen im Dezember 2020) 10 Ziobro J, Shellhaas R: Neonatal seizures: diagnosis, etiology, and management. Semin Neurol 2020; 40: 246-56 11 Sharpe C: Levetiracetam versus phenobarbital for neonatal seizures: a randomized controlled trial. Pediatrics 2020; 145: e20193182 (Erratum in: Pediatrics 2021; 147(1): e2020036806) 12 El-Dib M, Soul JS: The use of phenobarbital and other anti-seizure drugs in newborns. Semin Fetal Neonatal Med 2017; 22: 321-7 13 Bahi-Buisson N et al.: Epilepsies néonatales et erreurs innées du métabolisme. Arch Pediatr 2006; 13: 284-92
Das könnte Sie auch interessieren:

Diagnose der ALS: von Biomarkern bis Kognition und Verhalten
Die Heterogenität der ALS macht eine Diagnose nicht leicht. Dazu kommt, dass die Pathogenese immer noch nicht richtig verstanden ist und dass sich die Frühsymptome mit jenen anderer ...

Evidenzbasierte Ernährungsempfehlungen für ALS-Patient:innen
Mit ihrer progredienten neurodegenerativen Natur geht die ALS häufig mit erheblichen Herausforderungen im Ernährungsmanagement einher. Um evidenzbasierte Empfehlungen zu erarbeiten, ...

ALS: verbesserte Überlebensraten durch enterale Ernährung
Bei einem progressiven Verlauf der ALS mit Schluckbeschwerden kann eine Ernährungsunterstützung – z.B. mit PEG-Sonde – die Lebensqualität entscheidend verbessern. Eine aktuelle Studie ...