Kardiomyopathien bei neuromuskulären Erkrankungen
Neuromuskuläre Erkrankungen zeigen oft eine kardiale Mitbeteiligung mit Herzinsuffizienz und malignen Rhythmusstörungen. Durch frühzeitige Diagnostik und zeitgemäße Medikamenten- und Devicetherapie kann das Überleben trotz Mobilitätseinschränkung qualitativ hochwertig verlängert werden.
Keypoints
-
Die erste kardiologische Untersuchung soll gleich nach Diagnosestellung erfolgen.
-
Eine Myokardfibrose ist im MRT häufig auch bei normaler Linksventrikelfunktion nachweisbar.
-
Der Beginn einer Herzinsuffizienztherapie bei noch normaler Linksventrikelfunktion kann Symptomatik und Überleben verbessern.
-
Muskulär asymptomatische Konduktorinnen können ebenfalls Zeichen einer Kardiomyopathie aufweisen und sollen kardiologisch behandelt werden („female dystrophinopathy“).
Neuromuskuläre Erkrankungen (NMD) sind eine vielfältige Gruppe von Erkrankungen, die Motoneuronen, periphere Nerven, Synapsen oder die Muskelfasern selbst betreffen. Sie sind genetisch bedingt, meist fortschreitend und führen zu Muskelschwäche, Koordinationsstörungen, Immobilität und verkürzter Lebenserwartung. Während sich viele NMD bereits im Kindesalter klinisch manifestieren, werden manche jedoch erst spät symptomatisch. Andere Organfunktionen können dabei direkt oder indirekt betroffen sein wie die Atmung, die Verdauung, der Knochenstoffwechsel und das Herz-Kreislauf-System. Bei Letzterem können sich Kardiomyopathien (CMP) sowohl durch Rhythmusstörungen verschiedener Art als auch durch eine Herzinsuffizienz manifestieren. CMP und Herzinsuffizienz werden im klinischen Alltag oft synonym eingesetzt, sind aber nicht zwingend das Gleiche. Bei einer CMP kommt es zu strukturellen Abnormitäten des Myokards, die ihre Ursache nicht in Krankheiten wie Atherosklerose, arterieller Hypertonie oder in angeborenen bzw. erworbenen Vitien haben.1 Dabei haben die kardiale Morphologie und Gewebecharakterisierung einen hohen phänotypischen, aber nicht zwingend einen diagnostischen Stellenwert.
Eine Herzinsuffizienz hingegen ist unabhängig von der Ursache ein klinisches Syndrom mit typischen Symptomen (wie Atemnot bei Belastung und später auch in Ruhe, Schwäche oder Müdigkeit) und Zeichen (wie Tachykardie, pulmonale Rasselgeräusche oder periphere Ödeme) mit Nachweis einer strukturellen oder funktionellen Abnormität des Herzens.2
Kardiale Diagnostik
Das üblicherweise erste kardiale Bildgebungsverfahren ist die Echokardiografie, die die Morphologie des Herzens und die Pumpleistung der Ventrikel untersucht. Klassischerweise ist die systolische Funktion, die Auswurfleistung des Herzens („ejection fraction“, EF), beeinträchtigt („heart failure with reduced ejection fraction“, HFrEF). Jedoch können die klinischen Befunde einer Herzinsuffizienz auch auf Basis erhöhter Füllungsdrücke bei diastolischer Funktionsstörung und normaler systolischer Linksventrikelfunktion (LVF), also erhaltener Auswurfleistung, auftreten („heart failure with preserved ejection fraction“, HFpEF). Da bei zahlreichen Muskelerkrankungen eine körperliche Belastung, die zu Dyspnoe führen würde, gar nicht möglich ist, müssen echokardiografische und laborchemische Befunde (z.B. „N-terminal pro-B-type natriuretic peptide“, NT-pro-BNP) frühzeitig auch bei klinischer Beschwerdefreiheit erhoben werden, da sie von therapeutischer Konsequenz sind.
Durch kardiale Magnetresonanztomografie (cMRT) kann neben einer meist besseren Darstellung des Herzens (bei häufig infolge der neuromuskulären Skoliose eingeschränkter Ultraschallqualität) das Myokard genauer charakterisiert werden. Die Anreicherung des paramagnetischen Kontrastmittels Gadolinium als heller Anteil im ansonsten schwarzen Myokard („late gadolinium enhancement“, LGE) zeigt eine diffuse oder fokale Herzmuskelschädigung (z.B. Fibrose, Narbe, Entzündung), die oft typische Lokalisationen aufweist (z.B. posterolateral bei Dystrophinopathien, siehe Abb. 1, oder mittseptal bei Laminopathien). Das extrazelluläre Volumen (ECV), das bei NMD durch diffusen bindegewebigen Umbau im Myokard oft erhöht ist, kann ebenfalls errechnet werden.
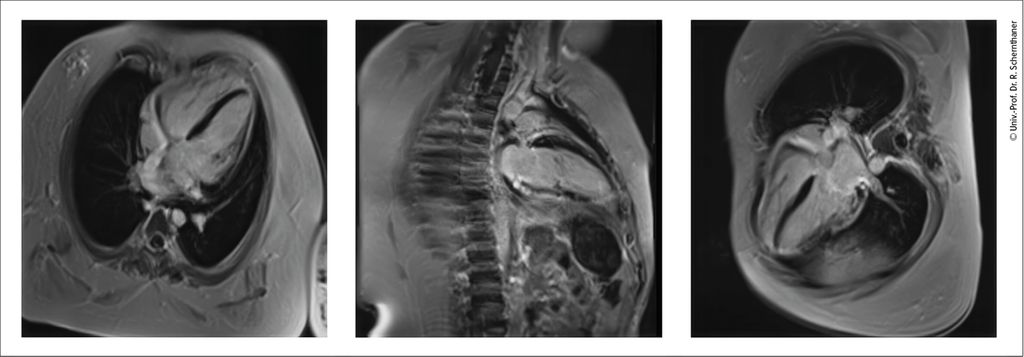
Abb. 1: Umschriebenes „late gadolinium enhancement“ apikal anterolateral, apikal posterolateral und apikal inferior sowie diffuse kleinfleckige Myokardfibrose in den lateralen Wandabschnitten bei einem 17-jährigen Patienten mit Muskeldystrophie Duchenne in der MRT (4-Kammer-, 2-Kammer-, 3-Kammer-Blick, Siemens Skyra 3T) (mit freundlicher Genehmigung des Zentralen Radiologie Instituts Klinik Landstraße, Wien)
Des Weiteren können Rhythmusstörungen bzw. Störungen des Reizleitungssystems auftreten. So finden sich bei myotoner Dystrophie gehäuft AV-Blockierungen unterschiedlicher Ausprägung (verlängertes PQ-Intervall bis hin zur völligen atrioventrikulären Dissoziation), Repolarisationsstörungen (ST-Streckendeviation) oder Schenkelblockbilder. Bei Dystrophinopathien wie Duchenne-Muskeldystrophie (DMD) oder Becker-Muskeldystrophie (BMD) ist das PQ-Intervall oft sehr kurz und der Kammerkomplex schenkelblockartig verbreitert (Abb. 2). Vorhofflimmern tritt bei myotoner Dystrophie oder fazioskapulohumeraler Dystrophie häufig auf, ventrikuläre Tachykardien und Kammerflimmern treten bei Laminopathien, Emery-Dreifuss-Muskeldystrophie oder einigen Formen der Gliedergürteldystrophien auf.
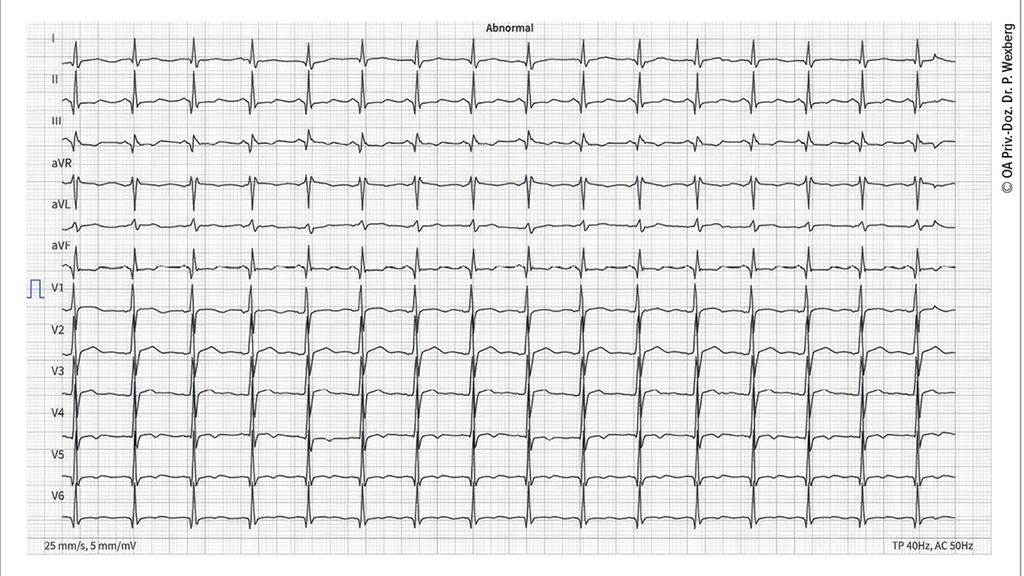
Abb. 2: EKG eines 16-jährigen Patienten mit Duchenne-Muskeldystrophie. Man beachte die Sinustachykardie (100/min), das verkürzte PQ-Intervall (113ms) und die Q-Zacken in I, II, III, aVF, V5-6
Die American Heart Association (AHA) empfiehlt daher nach Diagnosestellung einer NMD eine kardiologische Begutachtung mit klinischer Untersuchung, EKG und Herzultraschall.3 Für Erkrankungen mit einem erhöhten Risiko für Arrhythmien werden auch 24-Stunden-EKGs empfohlen. Bei Verdacht auf rhythmogene Schwindelzustände und Synkopen können die Implantation eines Looprecorders oder auch eine elektrophysiologische Herzkatheteruntersuchung diagnostische Klarheit bringen und kausale Therapien (Antiarrhythmika, Schrittmacher- oder Defibrillatorimplantation) ermöglichen. Je nach Diagnose und Symptomatik werden die Kontrollintervalle festgesetzt, wobei sich in der eigenen Erfahrung eine zumindest jährliche, besser halbjährliche Kontrolle gerade bei Dystrophinopathien und/oder reduzierter LVF als geeignet erwiesen hat. Abgesehen vom Erkennen einer allfälligen Verschlechterung erleichtern die häufigeren Kontakte die notwendigen Anpassungen der Medikation.
Medikamentöse Therapie- und Leitlinienempfehlung
Diese unterscheidet sich im Wesentlichen nicht von der Therapie der Herzinsuffizienz anderer Genese. Die vier Säulen der aktuellen medikamentösen Herzinsuffizienztherapie zur Reduktion der Mortalität sind in Tabelle 1 dargestellt.4 Bei hochgradig reduzierter LVF (EF <35%) werden alternativ zu ACE-Hemmern bzw. ATII- Blockern auch Angiotensinrezeptor/Neprilysin-Inhibitoren (ARNI) empfohlen, mit Sacubitril/Valsartan als einzig am Markt verfügbarer Substanz. Zur Symptomminderung und Reduktion der Hospitalisation kommen auch Diuretika (bei Volumenüberlastung), Ivabradin (Frequenzkontrolle bei Sinusrhythmus), Digitalis (Frequenzkontrolle bei Vorhofflimmern) sowie Antikoagulanzien (Verhinderung von Thromboembolien bei Vorhofflimmern) zum Einsatz.
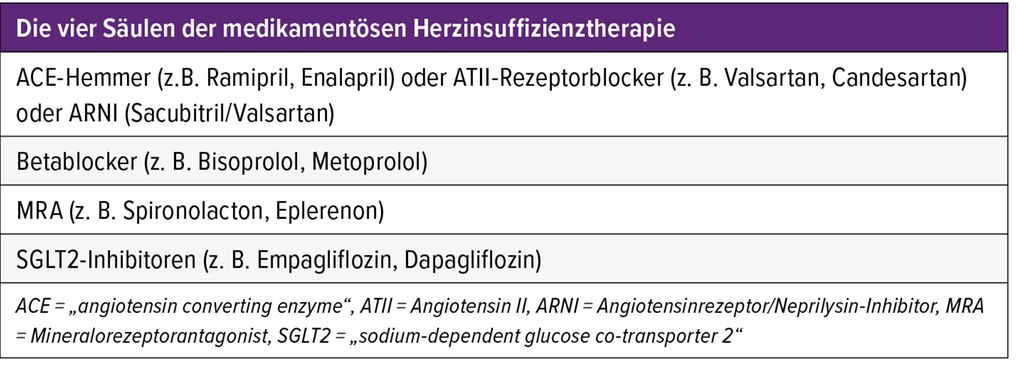
Tab. 1: Die vier Säulen der medikamentösen Herzinsuffizienztherapie (modifiziert nach McDonagh TA et al. 2023)4
Die europäischen Herzinsuffizienz-Guidelines wurden 2021 neu formuliert und haben 2023 angesichts des rasanten Therapiefortschritts ein Update erfahren. Hingegen wurden die einzig spezifischen NMD-Empfehlungen der AHA leider seit 2017 nicht mehr aktualisiert, weshalb sie gerade bei den Therapien wesentliche Neuerungen (in erster Linie hinsichtlich der SGLT2-Inhibitoren) nicht abbilden. In der klinischen Praxis empfiehlt es sich aber, auch hier nach allgemeinen kardiologischen Standards vorzugehen.
Bei allen NMD wird von der AHA bei reduzierter LVF der Einsatz von ACE-Hemmern empfohlen,3 bei DMD besteht ab einem Alter von 10 Jahren, aber auch bei normaler LVF eine Klasse-IIb-Empfehlung, da bereits bei ca. 60% der Erkrankten eine kardiale Mitbeteiligung vorliegt.5 Die meisten Therapiedaten liegen auch generell für DMD vor, können aber im Allgemeinen auf andere NMD übertragen werden. Die Behandlung mit ACE-Hemmern oder ATII-Blockern führt zu einer geringeren Sterblichkeit, einer langsameren Verschlechterung der LVF und der Myokardfibrose, vor allem wenn sie vor der Abnahme der LVF und dem Auftreten von Symptomen begonnen wird.6,7 Bei DMD wurde dies auch für MRA (Spironolacton, Eplerenon) gezeigt.3 Zudem hat die Kombination von ACE-Hemmern und Betablockern einen positiven Effekt auf die LVF bei DMD.7 Bei erhaltener EF werden Letztere aber nicht bzw. nur zur Behandlung von Arrhythmien empfohlen. Betablocker werden auch zur Frequenzkontrolle (Zielbereich bei Erwachsenen 70/min) und zur Prophylaxe lebensbedrohlicher ventrikulärer Tachykardien eingesetzt und finden somit bei Gliedergürteldystrophien, EDMD oder Laminopathien Verwendung. Bei Hypotonieneigung kann bei Sinusrhythmus auch der If-Kanalblocker Ivabradin eingesetzt werden, um die Herzfrequenz zu senken.8Zu SGLT2-Inhibitoren gibt es noch keine größeren Studien, sie werden aber bei Erwachsenen bereits bei NMD-Herzinsuffizienz eingesetzt. Bei Vorhofflimmern gilt die Indikation zur oralen Antikoagulation nach den gleichen Kriterien wie ohne NMD.
Devicetherapie
Außer der medikamentösen Therapie werden bei bradykarden Rhythmusstörungen Schrittmacher eingesetzt, wobei für Erkrankungen mit hohem Risiko für maligne ventrikuläre Rhythmusstörungen (wie Emery-Dreifuss, myotone Dystrophie1 oder Laminopathien) grundsätzlich großzügig die Indikation für einen implantierbaren Defibrillator (ICD) gestellt werden soll.3 Die individuelle Situation, der funktionelle Status und der Patient:innen-wunsch müssen ausführlich mit den Betroffenen diskutiert werden. Bei reduzierter Linksventrikelfunktion und Linksschenkelblock kann eine kardiale Resynchronisationstherapie durch Schrittmacher bzw. ICD mit einer zusätzlichen Sonde im Koronarsinus eingesetzt werden (Abb. 3). In Einzelfällen werden in Zentren mit der entsprechenden herzchirurgischen und anästhesiologischen Expertise „Kunstherzen“ („left ventricular assist devices“, LVAD) mit verhältnismäßig guten Langzeitüberlebensdaten implantiert.9,10
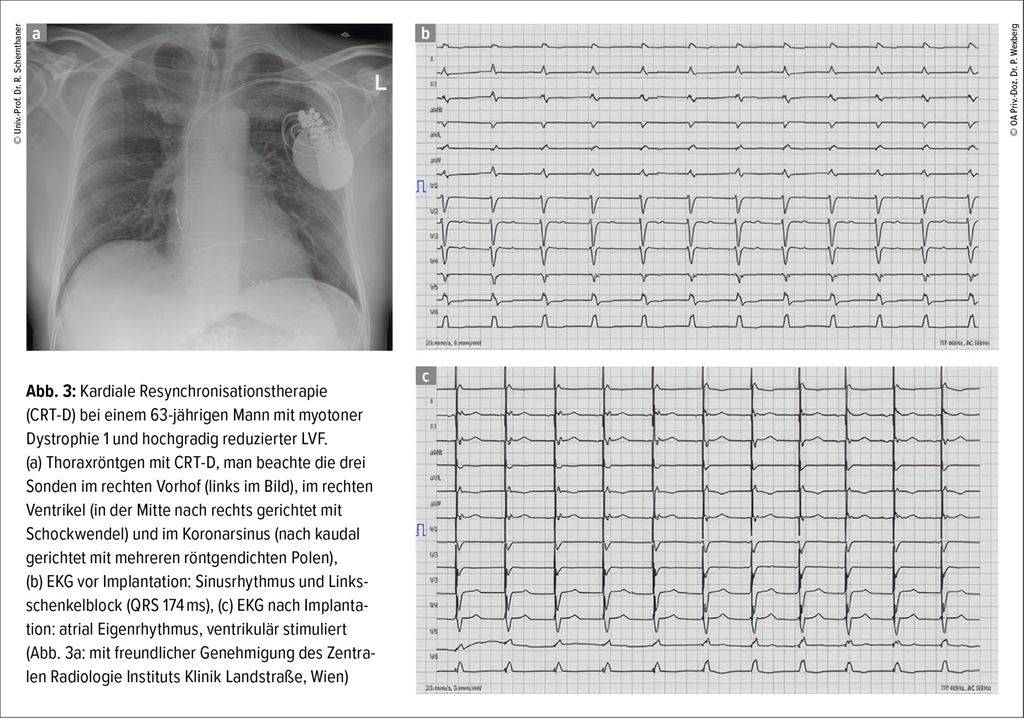
Abb. 3: Kardiale Resynchronisationstherapie (CRT-D) bei einem 63-jährigen Mann mit myotoner Dystrophie 1 und hochgradig reduzierter LVF. (a) Thoraxröntgen mit CRT-D, man beachte die drei Sonden im rechten Vorhof (links im Bild), im rechten Ventrikel (in der Mitte nach rechts gerichtet mit Schockwendel) und im Koronarsinus (nach kaudal gerichtet mit mehreren röntgendichten Polen), (b) EKG vor Implantation: Sinusrhythmus und Links-schenkelblock (QRS 174ms), (c) EKG nach Implanta-tion: atrial Eigenrhythmus, ventrikulär stimuliert (Abb. 3a: mit freundlicher Genehmigung des Zentralen Radiologie Instituts Klinik Landstraße, Wien)
Besonderheiten und Herausforderungen
Während die fortschreitende Mobilitätseinschränkung und die respiratorische Insuffizienz gerade bei den bereits im Kindesalter auftretenden NMD in der klinischen und psychosozialen Wahrnehmung im Vordergrund stehen, wird die kardiale Mitbeteiligung lange Zeit nicht als problematisch erlebt. Denn erstens wurde historisch bis vor Kurzem das Stadium der Herzinsuffizienz aufgrund der eingeschränkten Lebenserwartung bei vielen Patient:innen mit NMD oft gar nicht erst erreicht. Zweitens fehlt durch die eingeschränkte Mobilität die klinische Symptomatik. Und drittens stand bis vor wenigen Jahren noch nicht das ausgeprägte Armamentarium zur Behandlungen der Herzinsuffizienz zur Verfügung, wie wir es heute haben.11 Es ist daher oft herausfordernd, beispielsweise den Eltern eines Buben mit DMD die Notwendigkeit einer umfangreichen Medikation bei ihrem objektiv doch kardial asymptomatischen Sohn mit unauffälliger Echokardiografie verständlich zu machen. Gleichermaßen ist aber auch im medizinischen Bereich oft die Ansicht vertreten, dass eine kardiale Therapie angesichts der schweren Grunderkrankung nicht sinnvoll indiziert sei.
„Female dystrophinopathy“
Nicht vergessen werden darf, dass Konduktorinnen von Dystrophinopathien ebenfalls ein erhöhtes Risiko haben, eine Kardiomyopathie zu entwickeln („female dystrophinopathy“). Im eigenen Kollektiv fanden wir bei über 40% der untersuchten Frauen eine myokardiale Fibrose und ein erhöhtes myokardiales Extrazellulärvolumen.12 Hier ist in den letzten Jahren eine höhere Awareness entstanden, mit der Empfehlung, spätestens bei Nachweis einer myokardialen Fibrose eine Herzinsuffizienztherapie zu beginnen.13
Umgekehrt muss von kardiologischer Seite beachtet werden, dass Kortikosteroide, die im Allgemeinen mit einer Verschlechterung der LVF assoziiert werden, bei DMD günstig auf Muskelkraft, Lungenfunktion und auch die kardiale Funktion wirken und nicht „reflexartig“ abgesetzt werden sollen.
Fazit
Die Vielzahl der neuromuskulären Diagnosen, ihre klinische Mannigfaltigkeit und das immer längere Überleben erfordern eine enge Zusammenarbeit verschiedener Fachgebiete aus dem pädiatrischen und Erwachsenenbereich. Das oft schwere klinische Erscheinungsbild mit Immobilität, Muskelabbau und respiratorischer Insuffizienz darf nicht darüber hinwegtäuschen, dass neben den „Standards of Care“14–16 die aktuellen kardiologischen Therapiemöglichkeiten die Progredienz der Herzinsuffizienz verzögern, Arrhythmien bei NMD reduzieren und zu einer verbesserten Lebensqualität der Erkrankten führen.
Literatur:
1 Arbelo E et al.: 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J 2023; 44(37): 3503-626 2 McDonagh TA et al.: 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021; 42(36): 3599-726 3 Feingold B et al.: Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation 2017; 136(1): e200-31 4 McDonagh TA et al.: 2023 focused update of the 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2023; 44(37): 3627-39 5 Nigro G et al.: The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol 1990; 26(3): 271-7 6 Silva MC et al.: Myocardial fibrosis progression in Duchenne and Becker muscular dystrophy: a randomized clinical trial. JAMA Cardiol 2017; 2(2): 190-9 7 Conway KM et al.: Prophylactic use of cardiac medications and survival in Duchenne muscular dystrophy. Muscle Nerve 2025; 71(4): 574-82 8 Adorisio R et al.: Heart rate reduction strategy using ivabradine in end-stage Duchenne cardiomyopathy. Int J Cardiol 2019; 280: 99-103 9 Perri G et al.: Left ventricular assist device as destination therapy in cardiac end-stage dystrophinopathies: Midterm results. J Thorac Cardiovasc Surg 2017; 153(3): 669-74 10 Godfrey S et al.: Survival of the unfittest: the longest living LVAD-supported patient with DMD-associated cardiomyopathy. J Heart Lung Transplant 2023; 42(suppl): S360-1 11 Haddad CN et al.: Pharmacological management of dilated cardiomyopathy in Duchenne muscular dystrophy: a systematic review. Hellenic J Cardiol 2023; 74: 58-64 12 Wexberg P et al.: Myocardial late gadolinium enhancement is associated with clinical presentation in Duchenne muscular dystrophy carriers. J Cardiovasc Magn Reson 2016; 18(1): 61-8 13 Bourke J et al.: Cardiac care of children with dystrophinopathy and females carrying DMD-gene variations. Open Heart 2022; 9(2): e001977 14 Birnkrant DJ et al.: Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 2018; 17(3): 251-67 15 Birnkrant DJ et al.: Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 2018; 17(4): 347-61 16 Birnkrant DJ et al.: Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol 2018; 17(5): 445-55
Das könnte Sie auch interessieren:

Diagnose der ALS: von Biomarkern bis Kognition und Verhalten
Die Heterogenität der ALS macht eine Diagnose nicht leicht. Dazu kommt, dass die Pathogenese immer noch nicht richtig verstanden ist und dass sich die Frühsymptome mit jenen anderer ...

Evidenzbasierte Ernährungsempfehlungen für ALS-Patient:innen
Mit ihrer progredienten neurodegenerativen Natur geht die ALS häufig mit erheblichen Herausforderungen im Ernährungsmanagement einher. Um evidenzbasierte Empfehlungen zu erarbeiten, ...

ALS: verbesserte Überlebensraten durch enterale Ernährung
Bei einem progressiven Verlauf der ALS mit Schluckbeschwerden kann eine Ernährungsunterstützung – z.B. mit PEG-Sonde – die Lebensqualität entscheidend verbessern. Eine aktuelle Studie ...