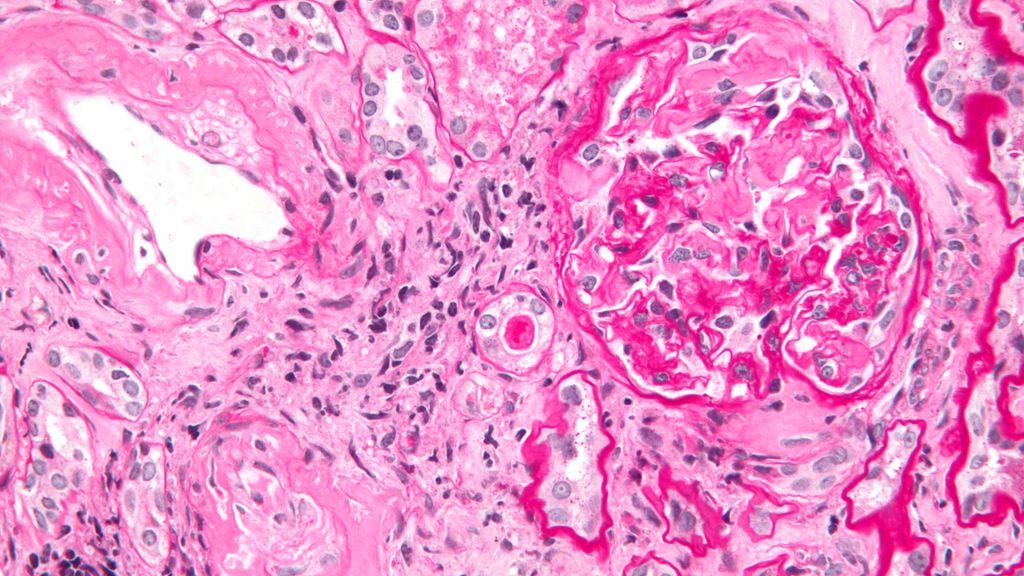
Renale Amyloidose
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die systemische Amyloidose ist eine heterogene Gruppe von Krankheiten, die sich durch Ablagerungen von falsch gefalteten Proteinen in verschiedenen Geweben auszeichnen. Es sind über 30 amyloidogene Proteine bekannt, wobei die Immunglobulin-Leichtketten, gefolgt von Transthyretin und Serumamyloid A die häufigsten sind. Die systemische Amyloidose gehört mit einer Inzidenz von circa 10 Patienten pro 1Mio. Einwohner zu den seltenen Krankheiten.1 Im Rahmen des Nephrologie Update Refreshers gab Prof. Dr. med. Thomas Fehr, Chefarzt Innere Medizin, Kantonsspital Graubünden, Chur, ein Update zu den Amyloidoseformen mit renaler Beteiligung.
Eine Beteilung der Nieren findet man am häufigsten bei der AL(Leichtketten)- und der AA(Amyloid A)-Amyloidose. Beide Formen können sich in allen Kompartimenten der Niere manifestieren.2Eine Nierenbeteiligung bei der ATTR(Transthyretin)-Amyloidose wird in der Literatur zwar beschrieben, ist aber sehr selten. Die renale Amyloidose äussert sich klinisch mit einer Proteinurie, häufig bis hin zu einem nephrotischen Syndrom und mit einer progressiven Verschlechterung der Nierenfunktion.
AA-Amyloidose
Die AA-Amyloidose (AAA) mit Ablagerungen von Amyloidfibrillen, die Serumamyloid A (SAA) enthalten, wird in der Regel durch chronische Entzündungszustände (autoimmun, infektiös, genetisch) verursacht. Das Serumamyloid A ist ein Akutphaseprotein, das in der Leber synthetisiert wird. Risikofaktoren der idiopathischen AAA sind das Alter, ein hoher BMI (Adipositas als inflammatorischer Zustand) sowie gewisse SAA1-Polymorphismen.3 Neben der Niere können auch die Leber mit einer Hepatosplenomegalie und der Darm mit einer Diarrhö (Proteinverlust-Enteropathie) sowie seltener Herz, Lunge und Schilddrüse befallen sein.
Histologisch lassen sich die SAA-Ablagerungen mittels Kongorot- oder immunhistochemischer Färbung nachweisen. Weitere diagnostische Optionen sind die 123I-SAP-Szintigrafie mit einer Sensitivität von 100%, die aber nur in wenigen Zentren zur Verfügung steht, und die PET-CT (und evtl. PET-MRT) zum Nachweis von Entzündungsherden.
Grosse Register zeigen, dass die Inzidenz der AAA stetig abnimmt und das Alter der betroffenen Patienten zum Zeitpunkt der Diagnose immer höher ist.4 «Der wichtigste Grund für diese Entwicklung, dürften die besseren Therapien sein, die uns zur Behandlung der chronisch-entzündlichen Krankheiten insbesondere aus dem rheumatologischen Formenkreis zur Verfügung stehen», sagte Fehr.
Therapie
Therapeutisch steht im Vordergrund die Behandlung der entzündlichen Grundkrankheit, Therapieziel ist die Normalisierung des SAA-Spiegels. Bei Patienten mit rheumatischen Erkrankungen und AAA konnte in verschiedenen Studien gezeigt werden, dass die erfolgreiche Behandlung der Grunderkrankung mit Zytokin-hemmenden Biologika auch mit einer deutlichen Verbesserung der renalen Symptome einhergeht.5–7
Eine spezifische Therapie für die AAA gibt es nicht. Vor 15 Jahren setzte man grosse Hoffnungen auf Eprodisat, ein leichtmolekulares Protein, das SAA bindet. In einer randomisierten Studie konnte die Abnahme der Nierenfunktion mit einer Eprodisatbehandlung signifikant verlangsamt werden. Auf harte Endpunkte wie Proteinurie, das Auftreten eines terminalen Nierenversagens und Tod konnte jedoch kein Effekt nachgewiesen werden, weshalb Eprodisat nie auf den Markt gekommen ist.8
AL-Amyloidose
Die Ursache der AL-Amyloidose (ALA) ist ein maligner Plasmazellklon, der Immunglobinketten produziert, die in verschiedenen Geweben abgelagert werden. Für eine monoklonale Lymphoproliferation mit mindestens einer renalen Läsion, die durch die Produktion des Immunglobulins verursacht wird, wird seit 2012 der Begriff der «monoclonal gammopathy with renal significance» (MGRS) verwendet. Der zugrunde liegende maligne B- oder Plasmazellklon verursacht dabei keine Tumorkrankheit und die hämatologischen Kriterien für eine spezifische Therapie sind nicht erfüllt. Die Indikation für eine allfällige Behandlung ist der Organbefall.9 Die MGRS umfasst derzeit ein Dutzend verschiedener Entitäten, wobei die am häufigsten vorkommende die Immunglobulin-Leichtketten(AL)-Amyloidose ist.9,10
Klinisch manifestiert sich die ALA mit den pathognomonischen periorbitalen Einblutungen (Waschbärenaugen) sowie Zeichen der Nieren- und Herzbeteiligung. Prognose und Schweregrad der renalen Erkrankung können anhand der Palladini-Klassifikation, die auf der 24h-Proteinurie und der GFR basiert, ermittelt werden. Für die kardiale Beteiligung gibt es eine Klassifikation der Mayo Clinic.11
Therapie
«Hinsichtlich der Therapie hat die ANDROMEDA-Studie das Bild entscheidend verändert»,12 so Fehr. In der randomisiert kontrollierten Studie wurde bei 388 Patienten mit ALA Daratumumabs.c. als Add-on zur bisherigen Standardtherapie aus Bortezomib, Cyclophosphamid und Dexamethason (VCD) verglichen mit VCD allein. Der Anteil der Patienten, die den primären Endpunkt komplette hämatologische Remission erreichten, war in der Daratumumab-Guppe signifikant grösser als in der Kontrollgruppe (53,3% vs. 18,1%) und auch das kardiale und renale Ansprechen war unter Daratumumab signifikant besser als unter VCD allein (41,5% vs. 22,2% resp. 53,0% vs. 23,9%) (Abb. 1).12
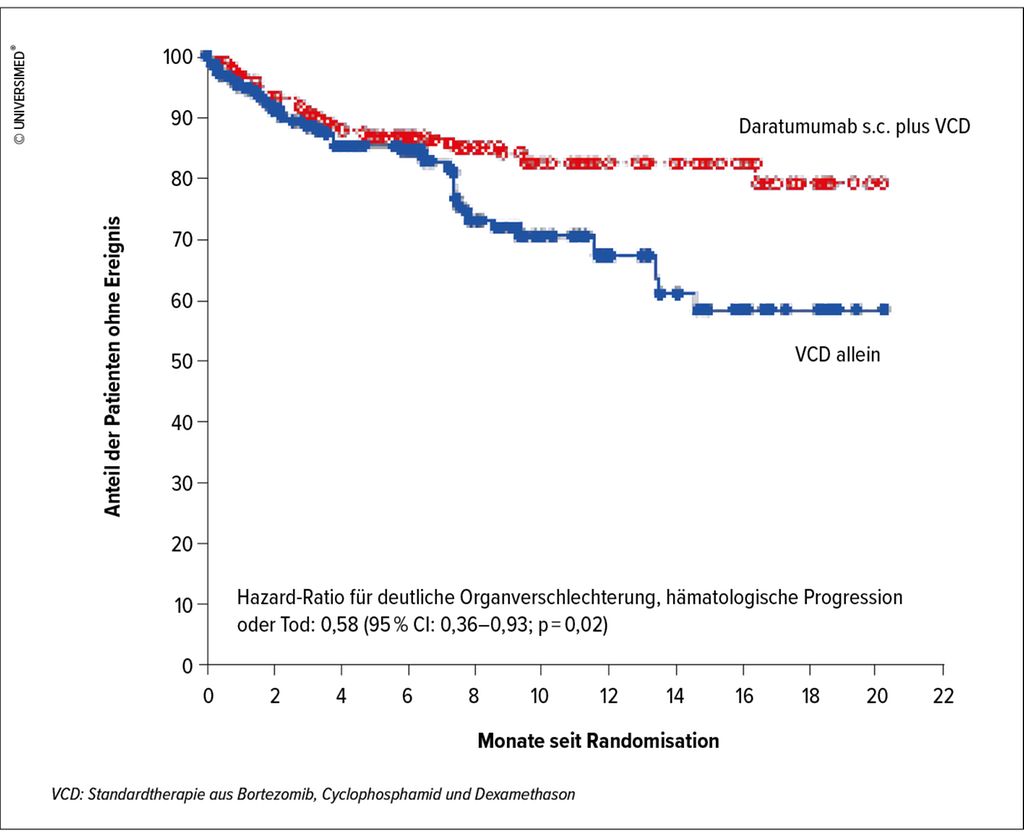
Abb. 1: Kaplan-Meier-Kurve des Überlebens ohne Verschlechterung der Organfunktion und ohne hämatologische Progression (modifiziert nach Kastritis et al.)12
Swiss Amyloidosis Network (SAN)
2013 wurde am USZ das Zürcher Amyloidoseregister gegründet, aus dem mit dem Anschluss zahlreicher weiterer Schweizer Zentren inzwischen das Schweizer Amyloidose-Netzwerk entstanden ist.
Im Register werden seit 2013 alle Amyloidosefälle prospektiv erfasst, zudem gibt es retrospektive Daten von 2005 bis 2013. Das Register enthält inzwischen Daten von rund 200 Patienten. Mit 50% ist die AL-Amyloidose die häufigste Form, gefolgt von der ATTR-Amyloidose mit 40% sowie der AA-Amyloidose und Fällen mit ungekanntem Typ mit je 5%. Bei der Inzidenz ist von 2005 bis 2020 eine deutliche Zunahme der ATTR-Amyloidose zu verzeichnen; im Zeitraum von 2016 bis 2020 war sie erstmals höher als die Inzidenz der AL-Amyloidose. Gleichzeitig zeichnet sich ein deutlicher Rückgang der Neuerkrankungen mit AA-Amyloidose ab. Das 5-Jahres-Gesamtüberleben liegt bei der AL- und der ATTR-Amyloidose bei circa 50%.13
Quelle:
FOMF Nephrologie Update Refresher, 24. und 25. Juni 2022, Zürich
Literatur:
1 Schwotzer R et al.: Expert recommendation from the Swiss Amyloidosis Network (SAN) for systemic AL-amyloidosis. Swiss Med Wkly 2020; 150: w20364 2 Gurung R, Tingting L: Renal amyloidosis: presentation, diagnosis, and management. Am J Med 2022; 135 (Suppl 1): S38-43 3 Blank N et al.: Obesity is a significant susceptibility factor for idiopathic AA amyloidosis. Amyloid 2018; 25: 37-45 4 Lane T et al.: Changing epidemiology of AA amyloidosis: clinical observations over 25 years at a single national referral centre. Amyloid 2017; 24: 162-6 5 Papa R, Lachmann HJ: Secondary, AA, Amyloidosis. Rheum Dis Clin North Am 2018; 44: 585-603 6 Ugurlu S et al.: Tocilizumab in the treatment of twelve cases with aa amyloidosis secondary to familial mediterranean fever. Orphanet J Rare Dis 2017; 12: 105 7 Lane T et al.: Safety and efficacy of empirical interleukin-1 inhibition using anakinra in AA amyloidosis of uncertain aetiology. Amyloid 2017; 24: 189-93 8 Dember LM et al.: Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med 2007; 356: 2349-60 9 Leung N et al.: The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol 2019; 15: 45-59 10 Yong ZH et al.: Kidney histopathologic spectrum and clinical indicators associated with MGRS. Clin J Am Soc Nephrol 2022; 17: 527-34 11 Dittrich T et al.: Prognosis and staging of AL amyloidosis. Acta Haematol 2020; 143: 388-400 12 Kastritis E et al.: Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med 2021; 385: 46-58 13 Schwotzer et al.: unpublished data 2022
Das könnte Sie auch interessieren:
Nephrokalzinose, Mutationen, Nierensteine – und was dies mit dem Alter zu tun hat
Prof. Martin Konrad leitet die Pädiatrische Nephrologie an der Universitätsklinik für Kinder- und Jugendmedizin in Münster. An der Jahrestagung der Schweizerischen Gesellschaft für ...
Aktuelles zu Nierensteinen, Lupusnephritis und chronischer Nierenkrankheit
Am Jahreskongress der Schweizerischen Gesellschaft für Nephrologie (SGN-SSN) in Interlaken stellten junge Forschungsteams im Rahmen der Posterpräsentationen ihre Ergebnisse vor. Wir ...
Adipositas und ihre Folgen für die Niere
Die Adipositas ist zu einem der wichtigsten weltweiten Gesundheitsprobleme geworden. Diese hat direkte und indirekte Folgen für die Niere. Neben der Gefahr einer Glomerulopathie und der ...