
Pneumopathie d’hypersensibilité: combien de fois les pathologistes, ne l’envisagent pas?
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
La pneumopathie d’hypersensibilité (PHS) est une pneumopathie interstitielle à médiation immunitaire qui est causée par des réactions immunitaires à des antigènes inhalés. Le diagnostic repose sur l’anamnèse, l’examen clinique, la radiologie, le lavage bronchoalvéolaire et, si nécessaire, une biopsie pulmonaire. Une caractéristique histologique centrale est la présence de granulomes non nécrosants. Toutefois, d’autres modifications pathologiques du parenchyme pulmonaire peuvent également suggérer une PHS, ce qui soulève la question suivante: combien de fois ne sont-elles pas détectées?
Keypoints
-
La pneumopathie d’hypersensibilité présente différentes modifications histopathologiques.
-
Elle reste une maladie sous-diagnostiquée, souvent en raison de problèmes lors du prélèvement d’échantillons.
-
La présence de granulomes n’est pas toujours nécessaire pour poser le diagnostic.
-
Un diagnostic précis est essentiel pour déterminer l’approche thérapeutique appropriée.
PHS en tant que pneumopathie interstitielle
Les pneumopathies interstitielles («interstitial lung disease», ILD) comprennent un groupe hétérogène de pneumopathies non néoplasiques, caractérisées par différents degrés d’inflammation et de fibrose.Ces affections touchent principalement les septa interalvéolaires, mais peuvent également impliquer les espaces aériens, certains présentant des caractéristiques morphologiques distinctives telles que des granulomes. Alors que de nombreuses ILD, comme les pneumonies atypiques, ont une cause infectieuse identifiable et un pronostic favorable, d’autres, dont les pneumopathies d’hypersensibilité et les pneumoconioses, sont liées à une exposition environnementale.
La PHS est une pneumopathie inflammatoire chronique et/ou fibrotique qui touche le parenchyme pulmonaire et les petites voies respiratoires. Elle est déclenchée par une réaction à médiation immunitaire à des antigènes inhalés, d’une taille généralement inférieure à 5μm, qui atteignent les alvéoles distales où ils provoquent une réaction inflammatoire. Selon les directives actuelles, la PHS est divisée en forme fibrotique et non fibrotique, une classification qui coïncide avec celle radiologique et qui a une importance pronostique. Le diagnostic d’une PHS reste complexe et nécessite une approche multidisciplinaire intégrant les résultats cliniques, radiologiques et histopathologiques.
Étiologie et épidémiologie de la PHS
La PHS est associée à plus de 50 expositions professionnelles et environnementales, dont des bactéries, des champignons, des protéines animales et des produits chimiques de bas poids moléculaire. L’étiologie varie en fonction de facteurs géographiques et professionnels. En Amérique du Nord, les antigènes aviaires et les moisissures sont des facteurs déclenchants fréquents, tandis que la PHS est associée aux climatiseurs contaminés par des moisissures dans les régions humides comme le Japon et l’Inde. La PHS peut survenir à tout âge et touche aussi bien les hommes que les femmes. Il est intéressant de noter que la prévalence est plus faible chez les fumeur·ses. Cela est dû à une diminution de la prolifération des lymphocytes et de l’activation des macrophages, ce qui affaiblit la réponse immunitaire aux antigènes inhalés.
L’identification et l’évitement des antigènes sensibilisants sont essentiels dans le diagnostic et le traitement. Cela s’avère toutefois souvent difficile, car aucune exposition spécifique ne peut être identifiée dans jusqu’à 60% des cas de PHS fibrotique. Cette incertitude diagnostique nécessite un examen clinique approfondi et une approche multidisciplinaire.
Tableau clinique de la PHS
La présentation clinique de la PHS est variable et est influencée par l’exposition aux antigènes, le degré de fibrose et la susceptibilité individuelle. Les symptômes sont en grande partie non spécifiques.
En cas de PHS non fibrotique, des symptômes précoces tels qu’une toux sèche, une dyspnée ainsi que des symptômes systémiques tels que de la fièvre, des frissons et un abattement surviennent, souvent liés dans le temps avec l’exposition aux antigènes. L’élimination des antigènes déclencheurs peut conduire à la résolution ou à la stabilisation de la maladie. Une exposition répétée peut cependant contribuer à la progression de la maladie.
La PHS fibrotique, quant à elle, se manifeste par une dyspnée à l’effort qui s’aggrave progressivement et une toux persistante.Par rapport à la forme fibrotique, la PHS non fibrotique a généralement un début aigu et une source antigène plus facilement identifiable.
Les tests de la fonction pulmonaire révèlent généralement un syndrome restrictif et une capacité de diffusion réduite. La recherche sérologique des anticorps IgG spécifiques à l’antigène n’est pas standardisée et présente une corrélation incohérente avec l’activité de la maladie. Les tests de provocation bronchique, bien qu’ayant une sensibilité et une spécificité modérées, sont rarement effectués dans la pratique clinique.
Caractéristiques radiologiques de la PHS
La tomodensitométrie haute résolution (TDM-HR) joue un rôle crucial dans le diagnostic de la PHS et dans la distinction entre les formes non fibrotique et fibrotique. La PHS non fibrotique se caractérise par des opacités bilatérales en verre dépoli, des nodules centrolobulaires et une perfusion en mosaïque avec piégeage d’air, particulièrement bien visibles sur les images à l’expiration. Une caractéristique distinctive est le «Three-Density Sign», un mélange de zones à densité élevée, normale et faible qui est hautement évocateur d’une PHS.
La PHS fibrotique présente les résultats suivants à l’imagerie: réticulation, bronchectasies de traction ou bronchectasies ainsi qu’«honeycombing» (aspect en nid d’abeille), souvent accompagnés de nodules centrolobulaires, de perfusion en mosaïque et de piégeage d’air, ce qui indique une obstruction bronchiolaire.Une caractéristique importante permettant de la distinguer de l’«usual interstitial pneumonia» (UIP) en cas de fibrose pulmonaire idiopathique (FPI) est que la PHS fibrotique peut ne pas toucher les bases pulmonaires ou peut présenter une prévalence dans les champs pulmonaires moyens à supérieurs. Cependant, certains cas de PHS fibrotique peuvent être similaires à l’UIP/FPI à l’imagerie, ce qui rend le diagnostic plus difficile. Alors que les résultats typiques de la TDM-HR soutiennent fortement le diagnostic de PHS, ils doivent toujours être interprétés dans le contexte de l’anamnèse clinique, de l’évaluation de l’exposition et des autres examens diagnostiques.
Évaluation pathologique et défis diagnostiques
La PHS non fibrotique présente typiquement une inflammation interstitielle chronique bronchiolocentrique, le parenchyme pulmonaire plus éloigné n’étant généralement pas touché. L’infiltrat est principalement constitué de lymphocytes, avec parfois des plasmocytes, une prédominance de plasmocytes pouvant indiquer un diagnostic alternatif tel qu’une «connective tissue disease-associated interstitial lung disease» (CTD-ILD; pneumopathie interstitielle associée à des connectivites). Les agrégats lymphoïdes sont généralement rares ou absents, et les centres germinatifs marqués sont inhabituels. Des granulomes ou des cellules géantes multinucléées apparaissent souvent dans l’interstitium péribronchiolaire et peuvent parfois contenir des cristaux de cholestérol ou des corps de Schaumann. Les granulomes de PHS ne présentent généralement pas la fibrose concentrique fine qui caractérise la sarcoïdose. En l’absence de granulomes, le résultat histologique peut ressembler à celui d’une pneumopathie interstitielle non spécifique (PINS) de type cellulaire, ce qui rend le diagnostic encore plus difficile.
La PHS fibrotique a une présentation pathologique très variable, ce qui en fait l’une des formes de pneumopathies interstitielles les plus difficiles à diagnostiquer. Elle peut prendre la forme d’une fibrose péribronchiolaire, d’une PINS fibrotique ou avoir un motif similaire à l’UIP. Contrairement à l’UIP/FPI, la PHS fibrotique présente souvent une métaplasie péribronchiolaire, qui touche une proportion importante des bronchioles. Même si les granulomes ou les cellules géantes sont plus rares dans la PHS fibrotique, leur présence confirme le diagnostic. Des foyers de fibroblastes peuvent être présents, mais ils sont généralement moins nombreux qu’en cas d’UIP/FPI.
Quelques diagnostics différentiels et leurs caractéristiques pathologiques sont résumés dans le Tableau 1.
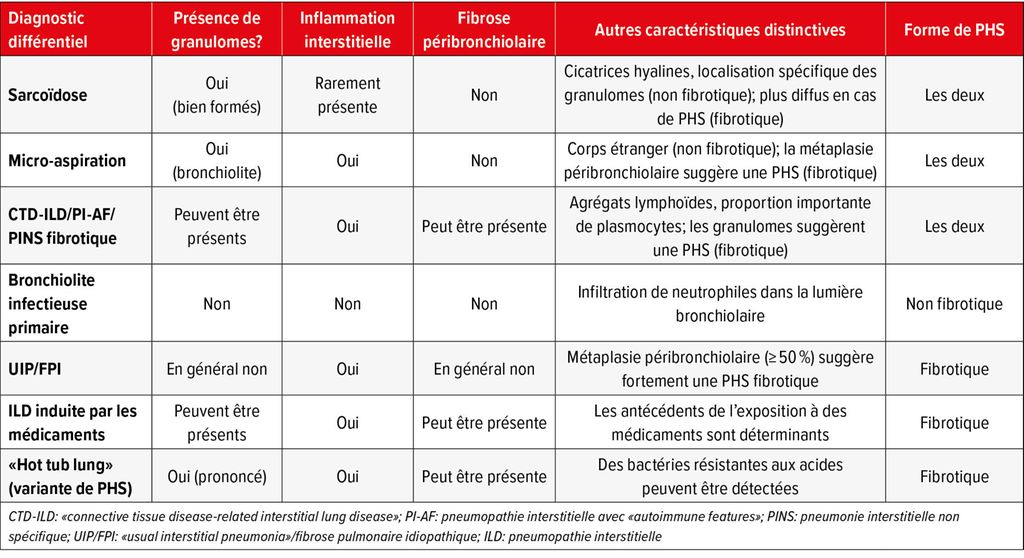
Tab.1: Diagnostics différentiels de la pneumopathie d’hypersensibilité (PHS)
Rôle de la biopsie et de la cryobiopsie transbronchiques dans le diagnostic de la PHS
La biopsie transbronchique (BTB) peut aider à diagnostiquer la PHS non fibrotique lorsqu’une inflammation interstitielle chronique associée à la présence de cellules géantes ou de granulomes est mise en évidence. L’inflammation interstitielle seule n’étant toutefois pas spécifique, le diagnostic final nécessite une corrélation avec les résultats cliniques et radiologiques.
La cryobiopsie transbronchique (CBTB) s’est imposée comme une alternative possible à la biopsie pulmonaire chirurgicale (BPC), car elle permet d’obtenir des échantillons tissulaires de taille plus importante sans artefacts de collapsus. Alors que la CBTB a montré une bonne concordance avec la BPC dans certaines études, des différences importantes ont été constatées dans d’autres, notamment pour distinguer la PHS fibrotique de l’UIP/FPI.Une limitation importante de la CBTB est la détection incohérente des granulomes et des cellules géantes, qui sont souvent uniquement répartis sous forme de taches. Des études montrent que la métaplasie péribronchiolaire, une caractéristique de la PHS fibrotique, peut être détectée de manière plus fiable dans les cryobiopsies – en particulier lorsque plusieurs échantillons sont prélevés. Il n’existe toutefois pas encore d’approche standardisée pour la CBTB et la variabilité de la taille des échantillons tissulaires influence la précision diagnostique. La BPC reste la méthode de référence dans le diagnostic de la PHS, en particulier dans les cas difficiles où la CBTB et les autres méthodes diagnostiques ne donnent aucun résultat clair. Bien que la BPC offre un meilleur résultat diagnostique, elle présente également des limites – notamment la variabilité du prélèvement et l’absence de granulomes ou de cellules géantes dans jusqu’à 30% des cas de PHS fibrotique. Malgré ces défis, les directives actuelles recommandent la BPC lorsque les méthodes non invasives telles que la TDM-HR et le lavage bronchoalvéolaire ne permettent pas de poser un diagnostic final.
La représentativité des échantillons tissulaires prélevés constitue un défi permanent pour toutes les procédures de biopsie.
Lavage bronchoalvéolaire (LBA)
Le LBA joue un rôle important dans le diagnostic, car il permet d’appréhender la réponse immunitaire en évaluant l’infiltration lymphocytaire médiée par les lymphocytes T. Une lymphocytose est plus prononcée en cas de PHS non fibrotique, tandis que la forme fibrotique présente souvent des valeurs plus faibles, inversement corrélées au degré de fibrose. Bien qu’elle puisse contribuer à exclure une PHS non fibrotique, l’absence de lymphocytose au LBA n’exclut pas la forme fibrotique. Un taux de lymphocytes de 40 à 60% au LBA, même s’il n’est pas spécifique, appuie fortement le diagnostic de PHS lorsqu’il est évalué conjointement avec les résultats cliniques et radiologiques. Une lymphocytose supérieure à 20% au LBA peut confirmer le diagnostic, en particulier dans le cas d’une PHS non fibrotique, mais elle n’est pas spécifique, car elle peut également être présente dans d’autres maladies telles que la sarcoïdose. En outre, le LBA aide à différencier la PHS d’autres pneumopathies interstitielles (ILD), comme la fibrose pulmonaire idiopathique (FPI), en particulier lorsqu’il est associé aux résultats de BTB, ce qui améliore la précision diagnostique.
Diagnostic multidisciplinaire de la PHS
Le diagnostic de la PHS nécessite une discussion multidisciplinaire incluant des corrélations cliniques, radiologiques et pathologiques (Fig.1). Un diagnostic peut souvent être posé sans biopsie en présence d’une anamnèse claire de l’exposition à un antigène connu et de résultats typiques de TDM-HR. Si l’anamnèse de l’exposition n’est pas claire ou les caractéristiques d’imagerie sont atypiques, une biopsie pulmonaire peut toutefois être nécessaire (Fig.1). L’interprétation des résultats de biopsie nécessite une corrélation minutieuse avec l’imagerie. Certain·es patient·es dont les images de TDM évoquent une PHS fibrotique peuvent présenter un motif de PINS ou d’UIP à la biopsie, tandis que d’autres avec des images de TDM compatibles avec un motif de PINS ou d’UIP peuvent présenter les caractéristiques histologiques d’une PHS. L’absence d’antigène connu est associée à un pronostic plus défavorable, ce qui souligne la nécessité d’une approche diagnostique globale prenant en compte toutes les informations cliniques, d’imagerie et histopathologiques disponibles.
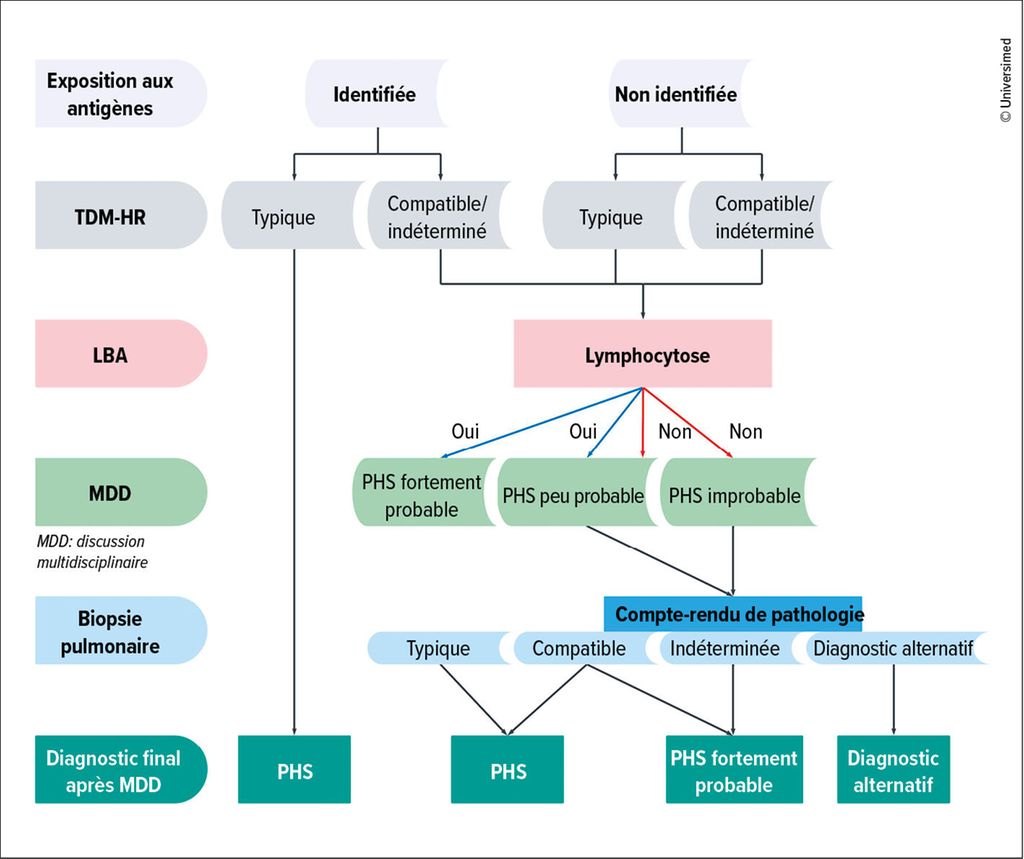
Fig.1: Le diagnostic de la pneumopathie d’hypersensibilité (PHS) nécessite une discussion multidisciplinaire incluant des corrélations cliniques, radiologiques et pathologiques
Littérature:
● Churg A: Hypersensitivity pneumonitis: new concepts and classifications. Modern Pathol 2022; 35: 15-27 ● Marinescu DC et al.: Integration and application of clinical practice guidelines for the diagnosis of idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Chest 2022; 162: 614-29 ● Moua T et al.: Challenges in the diagnosis and management of Fibrotic Hypersensitivity Pneumonitis: a practical review of current approaches. J Clin Med 2022; 11: 1473 ● Pérez ER et al.: Diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest 2021; 160: e97-156 ● Yang SR et al.: Diagnosis of hypersensitivity pneumonitis: review and summary of American College of Chest Physicians statement. Am J Surg Pathol 2022; 46: e71-93
Das könnte Sie auch interessieren:

Thérapies physiques en cas d’arthrite – nécessaires et utiles?
Le traitement des maladies articulaires rhumatismales inflammatoires a profondément évolué au cours des deux dernières décennies grâce à l’introduction de médicaments innovants: ...

Cancer du rein – stratégies actuelles et perspectives thérapeutiques futures
Les carcinomes rénaux non à cellules claires (non-ccRCC) sont pris en charge selon les mêmes standards que les carcinomes rénaux à cellules claires (ccRCC), mais les résultats ...

Chutes chez les personnes âgées: elles sont d’issue potentiellement fatale, mais aussi évitables
Les chutes chez les personnes âgées sont fréquentes et peuvent avoir de graves conséquences, mais elles ne sont souvent pas signalées par les personnes concernées en raison d’un ...