
Maladies plasmocytaires rares
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Le diagnostic précoce d’une gammapathie monoclonale de signification clinique (MGCS) est un défi dans la pratique clinique quotidienne en raison de l’hétérogénéité des manifestations et de sa rareté. Il est cependant essentiel pour le traitement précoce afin d’améliorer la fonction des organes et la survie.
Keypoints
-
LaMGCS regroupe les maladies dans lesquelles la protéine monoclonale produite par un petit clone de cellules B/plasmocytes provoque des lésions organiques ou tissulaires.
-
L’amylose AL est la maladie la plus fréquente et prototypique.
-
Le but du traitement est d’atteindre une rémission hématologique profonde qui permette d’améliorer la fonction des organes.
-
Une collaboration interdisciplinaire ainsi que l’inclusion dans des registres et des études cliniques sont essentielles.
Une gammapathie monoclonale de signification indéterminée (MGUS) peut être détectée chez trois pour cent des personnes âgées de plus de 50 ans.1
La majeure partie des personnes concernées ne présentent aucun symptôme et n’ont pas besoin de traitement. Le risque de progression vers un myélome multiple (MM) ou une maladie apparentée comme le lymphome lymphoplasmocytaire est de 1% par an.2
Si, en présence d’une gammapathie monoclonale, les symptômes suggèrent une lésion d’organe cible et ne peuvent être expliqués par aucune autre cause, il faut envisager le diagnostic rare de MGCS. Les signes d’alerte sont en particulier l’insuffisance cardiaque à fraction d’éjection ventriculaire gauche préservée (HFpEF), la détérioration de la fonction rénale, la protéinurie, la neuropathie et l’hépato-/splénomégalie, ainsi que l’apparition d’hématomes, de manifestations cutanées et d’œdèmes.3,4
Gammapathie monoclonalede signification clinique
LaMGCS regroupe un ensemble de maladies rares aux manifestations très hétérogènes dues à une lésion organique ou tissulaire. Celle-ci est provoquée par la protéine monoclonale produite par un petit clone agressif de cellules B/plasmocytes via différents mécanismes physiopathologiques, dont certains sont encore indéterminés. La protéine pathologique peut entraîner des lésions organiques et tissulaires par le biais de dépôts organisés ou non dans les organes et les tissus, par l’activité des auto-anticorps, par l’activation du système du complément par la voie alterne ainsi que par la médiation des cytokines (Tab.1 & 2).3–5
Chez les patient·es atteint·es de kératopathie cristalline, des dépôts cristallins d’immunoglobuline monoclonale peuvent être détectés dans la cornée.6
Les dépôts non organisés sont surtout présents dans la maladie des dépôts d’immunoglobuline monoclonale, qui est systémique et touche presque toujours les reins. Il s’agit souvent de dépôts de chaînes légères (maladie de dépôt des chaînes légères).7
Une activité des auto-anticorps dirigés contre la glycoprotéine associée à la myéline (MAG) peut entraîner une démyélinisation avec développement d’une neuropathie symétrique distale chronique, d’une ataxie sensorielle et de tremblements dans le cas d’une gammapathie monoclonale IgM.8
Une production accrue de VEGF joue un rôle clé dans la pathogenèse du syndrome POEMS. Il se caractérise par une polyneuropathie, une organomégalie, une endocrinopathie, une gammapathie monoclonale ainsi que des lésions cutanées, et s’accompagne typiquement de lésions osseuses ostéosclérotiques. Celles-ci peuvent être dues à une différenciation des ostéoblastes induite par le VEGF.9
La gammapathie monoclonale de signification rénale (MGRS) constitue un sous-groupe de laMGCS. Cette catégorie regroupe toutes les maladies dans lesquelles la protéine monoclonale sécrétée provoque une lésion rénale dans le cadre d’une gammapathie monoclonale. Cependant, les critères diagnostiques d’un MM, d’une maladie de Waldenström, d’une LLC ou d’un lymphome malin nécessitant un traitement ne sont pas remplis.10
On distingue les maladies limitées aux reins, comme la glomérulonéphrite à dépôts de C3 ou la glomérulopathie immunotactoïde, et les maladies systémiques avec atteinte rénale, comme l’amylose AL ou la maladie des dépôts d’immunoglobuline monoclonale. La réalisation d’une biopsie rénale et son traitement (microscopie électronique) dans un centre de référence sont essentiels pour poser le diagnostic deMGRS.10
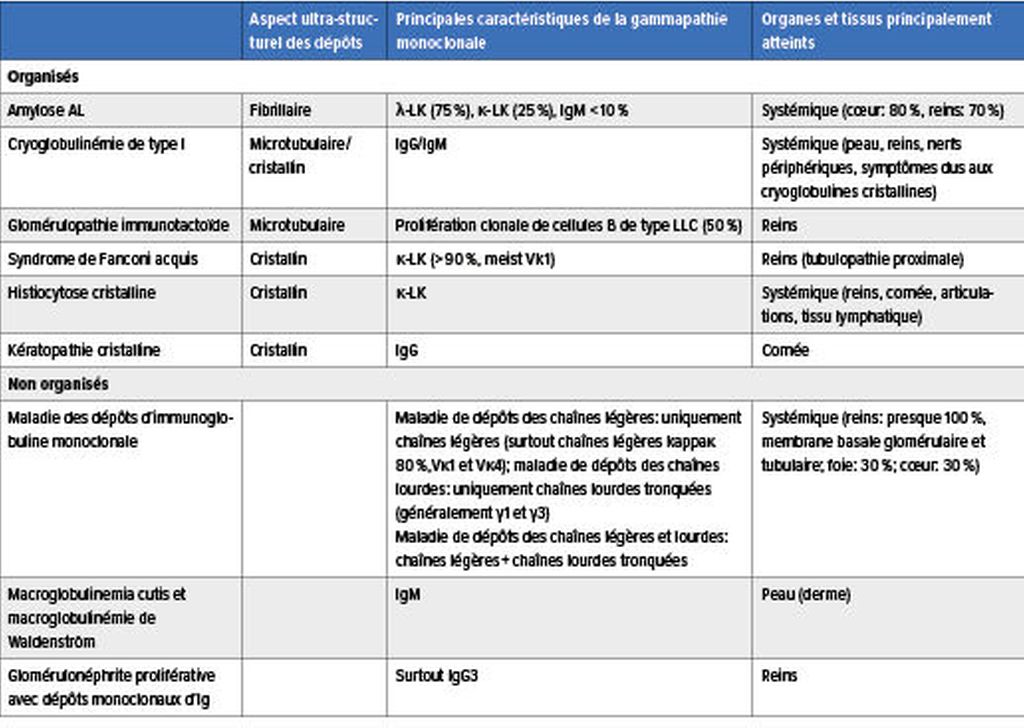
Tab.1: Mécanismes physiopathologiques de laMGCS: dépôts organisés et non organisés (modifié selon Fermand JP, 2018)3
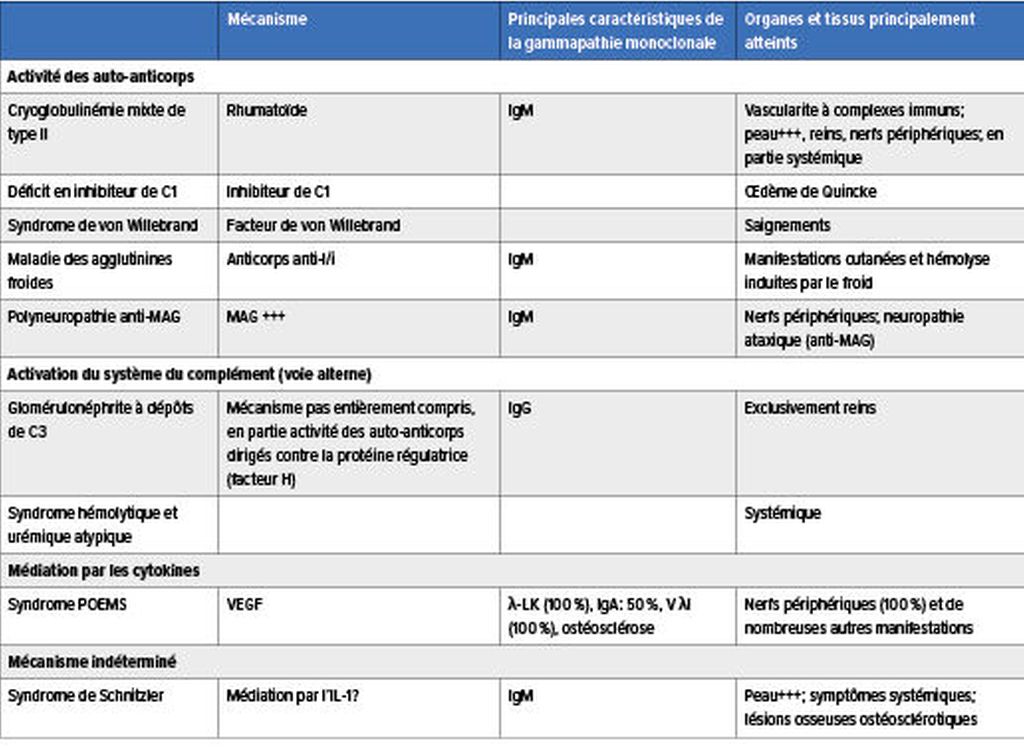
Tab.2: Mécanismes physiopathologiques de laMGCS: activité des auto-anticorps, activation du système du complément, médiation par les cytokines et mécanisme indéterminé (modifié selon Fermand JP, 2018);3 MAG: glycoprotéine associée à la myéline
Exclusion d’autres causes
En raison de la prévalence élevée de laMGUS, il est impératif d’exclure d’abord d’autres causes de lésions organiques ou tissulaires et d’établir un lien de causalité clair avec la gammapathie monoclonale. Les biopsies de l’organe concerné avec mise en évidence de dépôts d’immunoglobuline monoclonale pathologiques, qui doivent correspondre à la gammapathie monoclonale existante, jouent un rôle important à cet égard.3
En fonction de laMGCS attendue, il peut être nécessaire de procéder au dosage des titres d’auto-anticorps, des facteurs du complément et des cryoglobulines, ainsi qu’à un autre diagnostic par imagerie, analyse d’urine ou test de la coagulation. En outre, un diagnostic de la moelle osseuse doit être effectué pour caractériser le clone de cellules B/plasmocytes sous-jacent, car le traitement recommandé en dépend.3
En présence d’un clone lymphoplasmocytaire produisant des IgM (CD20+), des traitements à base d’anticorps monoclonaux anti-CD20 sont mis en place. En revanche, en présence d’un clone de plasmocytes produisant des IgG, IgA ou formant des chaînes légères (CD38+, CD138+, CD20–), on utilise des substances issues du traitement du MM, principalement le bortézomib en association avec des alkylants et de la dexaméthasone (CyBorD), des anticorps monoclonaux anti-CD38 ainsi que le traitement par le melphalan à haute dose en combinaison avec une autogreffe de cellules souches hématopoïétiques (HDT/ABSZT).3
Les immunoglobulines à haute dose et les corticoïdes sont utilisés dans le traitement de laMGCS d’origine auto-immune.3
Le but du traitement de laMGCS est d’obtenir une rémission hématologique associée à une amélioration de la fonction des organes.11
Une collaboration interdisciplinaire ainsi que l’inclusion dans des registres et des études cliniques sont essentielles pour les patient·es atteint·es deMGCS.
Amylose AL
L’amylose à chaînes légères (AL) est la maladie la plus fréquente et prototypique dans le groupe desMGCS. L’incidence est d’environ neuf personnes par million d’habitants/an.12
La sécrétion monotypique de chaînes légères lambda est nettement plus fréquente que celle de chaînes légères kappa. Sur le plan physiopathologique, un mauvais repliement des protéines entraîne le dépôt de fibrilles amyloïdes insolubles dans les tissus et les organes.13
Une atteinte avancée des organes, en particulier du cœur, est associée à un pronostic défavorable.14
Selon l’importance de l’atteinte du cœur ou des reins, en fonction de biomarqueurs définis (NT-proBNP, cTnT, BNP, DFGe, protéinurie), on peut distinguer différents stades avec des pronostics différents.14–16
Pour poser le diagnostic, il est nécessaire de procéder à une biopsie tissulaire avec mise en évidence des dépôts amyloïdes par coloration au rouge Congo. Pour distinguer l’amylose des autres formes, il faut en outre procéder à un typage de l’amylose. La méthode de référence est le séquençage, la méthode la plus pratiquée étant l’immunohistochimie.17
Le traitement de première intention de référence dans l’amylose AL est à base de daratumumab, cyclophosphamide, bortézomib et dexaméthasone (Daratumumab-CyBorD).18
Les patient·es éligibles pour un HDT/ABSZT par le melphalan reçoivent deux à quatre cycles de traitement d’induction. Les patient·es non éligibles à un HDT/ABSZT recevront six cycles de daratumumab-CyBorD suivis d’un traitement d’entretien par le daratumumab. Si, après quatre cycles de traitement par daratumumab-CyBorD, on n’obtient pas au moins une très bonne rémission partielle de la maladie, définie par une réduction jusqu’à <40mg/l de la différence entre la chaîne légère libre impliquée et celle qui ne l’est pas, la maladie est considérée comme réfractaire ou progressive. Il convient alors de changer de traitement. En principe, le but du traitement est d’atteindre une rémission hématologique profonde, nécessaire pour obtenir une amélioration de la fonction des organes et une prolongation de la survie.
Littérature:
1 Kyle RA et al.: Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 2006; 354(13): 1362-9 2 Kyle RA et al.: A long-term study of prognosis in monoclonal gammopathy of undetermined significance. NEngl J Med 2002; 346(8): 564-9 3 Fermand JP et al.: Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood 2018; 132(14): 1478-85 4 Moreno DF et al.: Treatment of patients with monoclonal gammopathy of clinical significance. Cancers 2021; 13(20): 5131 5 Merlini G, Palladini G: Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematol Am Soc Hematol Educ Program 2012; 2012: 595-603 6 Milman T et al.: Paraproteinemic keratopathy: the expanding diversity of clinical and pathologic manifestations. Ophthalmology 2015; 122(9): 1748-56 7 Cassano Cassano R et al.: Light chain deposition disease: pathogenesis, clinical characteristics and treatment strategies. Ann Hematol 2024; doi: 10.1007/s00277-024-05911-9 8 Khwaja J et al.: IgM monoclonal gammopathies of clinical significance: diagnosis and management. Haematologica 2022; 107(9): 2037-50 9 Dispenzieri A: POEMS syndrome: 2019 update on diagnosis, risk-stratification, and management. Am J Hematol 2019; 94(7): 812-27 10 Leung N et al.: The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol 2019; 15(1): 45-59 11 Cohen C et al.: Bortezomib produces high hematological response rates with prolonged renal survival in monoclonal immunoglobulin deposition disease. Kidney Int 2010; 88(5): 1135-43 12 Kyle R et al.: Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989 [see comments]. Blood 1992; 79(7): 1817-22 13 Palladini G et al.: Management of AL amyloidosis in 2020. Blood 2020; 136(23): 2620-7 14 Dispenzieri A et al.: Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. JClin Oncol Off J Am Soc Clin Oncol 2004; 22(18): 3751-7 15 Lilleness B et al.: Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood 2019; 133(3): 215-23 16 Palladini G et al.: Astaging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood 2014; 124(15): 2325-32 17 Schönland SO et al.: Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood 2012; 119(2): 488-93 18 Gertz MA: Immunoglobulin light chain amyloidosis: 2024 update on diagnosis, prognosis, and treatment. Am J Hematol 2024; 99(2): 309-24
Das könnte Sie auch interessieren:

État des lieux sur l’asthme: AIR, MART et médicaments biologiques
Les SABA, longtemps utilisés dans le traitement de l’asthme bronchique, sont aujourd’hui largement obsolètes et se sont révélés moins efficaces qu’un traitement combiné d’entretien et de ...
Souvent, une antibioprophylaxie n’est pas nécessaire
La question de savoir si une antibioprophylaxie est nécessaire lors des interventions endoscopiques, en particulier au niveau du tractus gastro-intestinal, fait toujours l’objet de ...
Médicaments spécifiques à l’ATTR-CM: revue systématique et méta-analyse
La présente méta-analyse1 est le premier travail qui harmonise les études cliniques randomisées contrôlées sur le traitement spécifique à l’amylose dans l’amylose à transthyrétine avec ...