Autoimmune Lebererkrankungen
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die primär biliäre Cholangitis (PBC), die Autoimmunhepatitis (AIH) und die primär sklerosierende Cholangitis (PSC) gehören zum Spektrum der seltenen Lebererkrankungen und stellen oftmals sowohl diagnostisch als auch therapeutisch eine Herausforderung dar.
Keypoints
-
Bei ca. einem Drittel der Patient*innen mit autoimmunen Lebererkrankungen besteht bereits zum Zeitpunkt der Erstdiagnose eine Leberzirrhose.
-
Von der primär biliären Cholangitis (PBC) sind vor allem Frauen (80%) betroffen.
-
Die Autoimmunhepatitis (AIH) kann sich in jedem Lebensalter erstmanifestieren und tritt bei Frauen deutlich häufiger auf (ca. 4:1).
-
Die primär sklerosierende Cholangitis (PSC) betrifft hingegen in erster Linie Männer (3:1), meist im Alter unter 40 Jahren, und geht in 70% der Fälle mit einer begleitenden chronisch-entzündlichen Darmerkrankung einher.
Das metabolische Syndrom, übermäßiger Alkoholkonsum und chronische Virushepatitiden sind weltweit für mehr als 95% der chronischen Lebererkrankungen verantwortlich. Aufgrund der geringeren Gesamtprävalenz rutscht die Vielfalt der seltenen Lebererkrankungen oftmals an den Rand unseres diagnostischen Wahrnehmungshorizontes. Dabei ist es gerade in diesem Patient*innenkollektiv besonders wichtig, rascheine Diagnose zu stellen, zeitgerecht eine Therapie zu etablieren und damit möglichst die Erkrankungsprogression in Richtung Leberzirrhose zu unterbinden. Insbesondere Vertreter der autoimmunologisch vermittelten Lebererkrankungen treten ab und zu in Begleitung rheumatologischer Systemerkrankungen auf und werden daher im Rahmen dieses Artikels diskutiert.
Die primär biliäre Cholangitis (PBC)
Die primär biliäre Cholangitis (PBC) ist eine autoimmun mediierte, cholestatische Lebererkrankung der kleinen bis mittelgroßen Gallengänge, die aufgrund genetischer Prädisposition und verschiedener Umweltfaktoren zu einem immunologischen Toleranzverlust gegenüber biliären Epithelzellen führt.1
Frauen sind vornehmlich von der PBC betroffen (in bis zu 80% der Fälle) und präsentieren sich meist mit einem profunden, insbesondere nächtlich auftretenden Juckreiz, gepaart mit einer protrahierten Fatigue. Laborchemisch zeigt sich eine Auslenkung der alkalischen Phosphatase, der gGT und ggf. des Bilirubins. Serologisch kann das Vorliegen antimitochondrialer Antikörper gegen den Pyruvat-Dehydrogenase-II-Komplex (AMA-M2) weitere diagnostische Hinweise liefern. Sollten AMA-M2-Antikörper nicht nachweisbar sein, kann das Vorliegen der ANA-Subsets SP100 und GP210 die Diagnose in die richtige Richtung führen. Insgesamt müssen mindestens zwei der drei Diagnosekriterien, also ausgelenkte Cholestaseparameter, positive Autoimmunserologie und/oder PBC-typische bioptische Veränderungen, nachweisbar sein, um die Diagnose einer PBC stellen zu können.
Ursodesoxycholsäure (UDCA; 13–15mg/kg KG), eine tertiäre Gallensäure, wird als First-Line-Therapie verschrieben, wirkt zytoprotektiv und fördert die biliäre Bikarbonatsekretion.1 Allerdings zeigen ca. 30–40% der Patient*innen hierauf kein adäquates Ansprechen (definiert als persistierende AST- oder AP-Erhöhung ≥1,5xULN und fehlende Bilirubinnormalisierung nach 6 Monaten Therapie [Paris-2-Kriterien]).2 Ein fehlendes UDCA-Ansprechen ist mit einer rascheren Krankheitsprogression und einem höheren Risiko für Dekompensationsereignisse (Varizenblutung, hepatische Enzephalopathie und Aszites), Lebertransplantationen und Mortalität assoziiert.3
Als Zweitlinientherapie kann daher additiv Obeticholsäure,4 ein Farnesoid-X-Rezeptor-Agonist, oder Bezafibrat (400mg/d), ein primär für die Hypertriglyzeridämie zugelassener PPAR/PXR-Agonist, verschrieben werden. Insbesondere für die Add-on-Therapie mit Bezafibrat konnte sowohl ein verbessertes biochemisches Ansprechen (30% [UDCA + BZF] vs. 1% [UDCA] nach 15 Monaten)5 als auch ein positiver Effekt auf das transplantatfreie Überleben gezeigt werden (67%ige Reduktion des gemeinsamen Endpunktes Tod/Lebertransplantation in der BZF+UDCA-Gruppe; HR: 0,3253 [95% CI:0,1936–0,5466; p<0,0001]).6 Trotz der genannten Therapiemöglichkeiten liegt das transplantatfreie Gesamtüberleben nach 10 Jahren bei 83,1%. Das Vorliegen einer fortgeschrittenen Lebererkrankung mit Zeichen der klinisch signifikanten portalen Hypertension (transplantatfreies 10-Jahres-Überleben: 57,4%), aber auch das Nichtansprechen auf die primäre UDCA-Therapie ist mit einer schlechteren Prognose assoziiert.3,7
Autoimmunhepatitis (AIH)
Mit einer Prävalenz von 15–25/1000008 gehört die Autoimmunhepatitis (AIH)ebenfalls in den Formenkreis der seltenen Lebererkrankungen. Ausgelöst durch Umweltfaktoren, wie z.B. Virusinfektionen, kommt es zu einem immunologischen Toleranzverlust mit Entstehung autoreaktiver CD4+- und CD8+-T-Lymphozyten, die sich gegen hepatozytäre Antigene richten.9

Abb.: Die rasche Diagnose und Therapie einer autoimmunen Lebererkrankung ist wichtig, um die Erkrankungsprogression in Richtung Leberzirrhose zu unterbinden
Die Erkrankung kann sich in jedem Lebensalter erstmanifestieren und betrifft Frauen deutlich häufiger als Männer (ca. 4:1)8. Die Palette der klinischen Beschwerden reicht von asymptomatischen Verläufen über unspezifische Beschwerden – wie Müdigkeit, Arthralgien und Oberbauchschmerzen – bis hin zu hochakuten Erstmanifestationen mit Koagulopathie, hepatischer Enzephalopathie und Ikterus im Sinne eines akuten Leberversagens (bis zu 6% der Fälle).8 Aufgrund des oft subklinischen Verlaufs mit dadurch verspäteter Diagnose kann bereits bei ca. einem Drittel der Patient*innen im Rahmen der Erstmanifestation eine kompensierte Leberzirrhose diagnostiziert werden.8
Ausgelenkte Transaminasen mit einem AST/ALT-Verhältnis (De-Ritis-Quotient) von <1 weisen auf ein hepatitisches Geschehen hin, geben jedoch naturgemäß noch keinen klaren Hinweis auf die zugrundeliegende Genese. Ausgelenkte Gammaglobuline bzw. erhöhte IgG-Spiegel stellen laborchemisch einen weiteren Hinweis dar.
In der Autoimmunserologie können antinukleäre Antikörper (ANA), Antikörper gegen glatte Muskelzellen (ASMA), Anti-LKM-1 und -3, Anti-LC-1, Anti-SLA/LP, pANCA und Ro52 nachweisbar sein, sind jedoch alleine oftmals nur unspezifisch.8 Ein weiteres diagnostisches Tool ist die Leberbiopsie, die durch Nachweis AIH-typischer histologischer Veränderungen, wie z.B. der Interface-Hepatitis, weitere wichtige Hinweise für die Diagnosefindung liefert. Um verschiedene diagnostische Parameter in die Diagnosefindung der AIH zu integrieren, wurde der IAIHG-Score der International Autoimmune Hepatitis Group entworfen, mit dessen Hilfe die Validität einer AIH-Diagnose abgeschätzt werden kann (Tab. 1).10
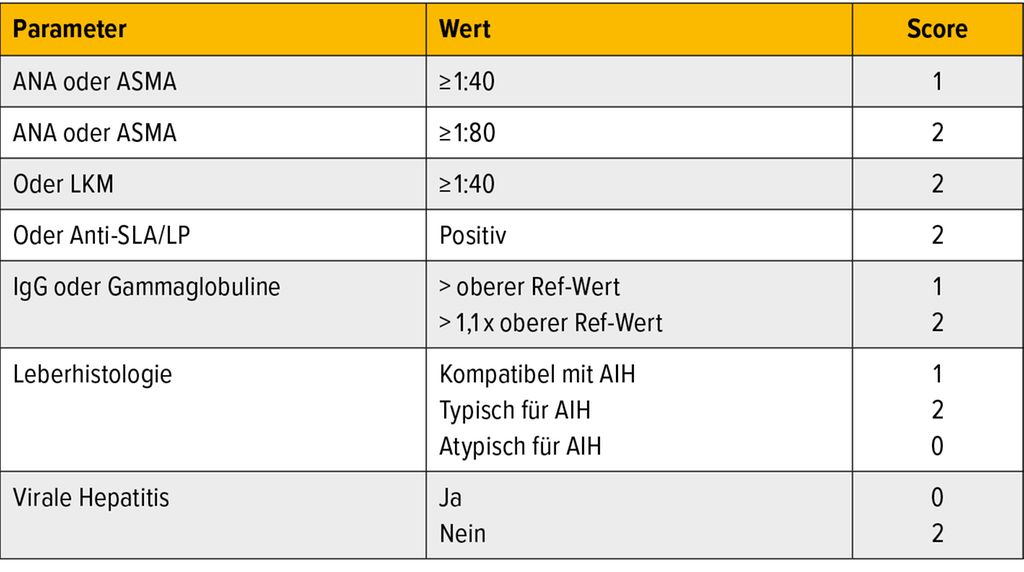
Tab. 1: IAIHG-Score: ≥7 definitive AIH-Diagnose; ≥6 wahrscheinliche AIH-Diagnose (modifiziert nach Hennes EM et al. 2008)10
Therapeutisch erhalten Patient*innen mit einer AIH eine Induktionstherapie mit einem oralen Kortikosteroid (1mg/kg KG) unter gleichzeitiger Immunsuppression mit Azathioprin. Primär sollten die Transaminasen innerhalb der ersten 2 Therapiewochen um 25% gesunken sein.8 Ab Normalisierung der Gammaglobuline und der ALT („biochemical response“) ist die Therapie für weitere 2 Jahre, jedoch mindestens für insgesamt 3 Jahre, fortzuführen, bevor ein Absetzversuch der immunsuppressiven Therapie gewagt werden kann (Relapsrisiko 50–90% in 12 Monaten).8 Bei Patient*innen, die bisher noch keine Leberzirrhose entwickelt haben, kann eine immunsuppressive Therapie auch mit Budesonid begonnen werden (9mg/d), wodurch Nebenwirkungen der systemischen Steroidtherapie umgangen werden.8 Unter Therapie haben Patient*innen mit AIH eine normale Lebenserwartung. Indikatoren für eine schlechtere Prognose sind eine verspätete Diagnose und eine hohe histologische Entzündungsaktivität.
Primär sklerosierende Cholangitis (PSC)
Die primär sklerosierende Cholangitis ist eine seltene (Inzidenz 1:200000),11 autoimmun mediierte, chronische Entzündung der intra- und extrahepatischen Gallenwege, die bei anhaltender Krankheitsaktivität zu fibrösen Stenosen der Gallenwege und damit zur Obstruktion des Gallenabflusses führt.12 Die PSC betrifft in erster Linie Männer (3:1), meist unter 40 Jahren,12 und geht in 70% der Fälle mit einer begleitenden chronisch-entzündlichen Darmerkrankung einher (meist mit einer Colitis ulcerosa)13.
Auffällig wird die PSC meist durch einen Anstieg der Cholestaseparameter.12,14Zu einem großen Anteil sind die Betroffenen zum Zeitpunkt der Diagnose noch asymptomatisch, allerdings entwickeln viele Patient*innen im weiteren Krankheitsverlauf rezidivierende Oberbauchschmerzen, Juckreiz, Ikterus und Müdigkeit.15 Eine deutliche Auslenkung der Transaminasen deutet oftmals auf eine begleitende Autoimmunhepatitis im Sinne eines sog. AIH-Overlap-Syndroms hin, die bis zu 14% der Patient*innen mit PSC betrifft.16 Ein deutlicher Bilirubinanstieg kann auf eine signifikante Gangstriktur oder aber auf ein cholangiozelluläres Karzinom (CCC) hinweisen – eine gefürchtete Langzeitkomplikation, an die insbesondere bei gleichzeitig bestehendem Fieber, Gewichtsverlust und Abdominalgie zudenken ist.12,16
Es gibt keine spezifischen Autoantikörper, die das Vorhandensein einer PSC bestätigen oder ausschließen können. pANCA finden sich in 80% der Fälle, sind allerdings relativ unspezifisch.12 Diagnostisch spielt die Magnetresonanz-Cholangiopankreatikografie (MRCP) eine große Rolle, mit der die typischen, perlenkettenschnurartigen Einschnürungen der Gallenwege oft gut visualisiert werden können (86% Sensitivität, 94% Spezifität). Die invasive Abklärung mittels ERCP bleibt Fällen mit invasiv-therapeutischer Intention oder persistierendem klinischem Verdacht, trotz negativer MRCP, vorbehalten.12,17 Von einer Leberbiopsie zur Abklärung der PSC wird zunehmend Abstand genommen, es sei denn, es besteht der Verdacht auf eine begleitende Autoimmunhepatitis oder eine Small-Duct-PSC (betrifft ca. 9% der Patient*innen).11, 17
Ursodesoxycholsäure (UDCA) ist die am weitesten verbreitete Therapieoption und wird auch weiterhin – trotz kontroversieller Guidelineempfehlungen18,19 und fehlenden therapeutischen Effekts auf klinisch signifikante Endpunkte (Überleben, Lebertransplantation)20 – in einer Dosierung von 13–15mg/kg KG verabreicht, da trotz allem in bisherigen Studien ein Rückgang der AP verzeichnet werden konnte.21
Populationsbasierte Studien bei kaukasischen PSC-Patient*innen mit flächendeckendem UDCA-Einsatz beobachteten ein 10-Jahres-Gesamtüberleben von 65%.22 Junges Alter bei Diagnose, weiblichesGeschlecht und Small-Duct-PSC sind mit einer besseren Prognose assoziiert, wohingegen Gallengangsstrikturen, wiederholte Cholangitiden, eine begleitende Colitis ulcerosa, Leberzirrhose und portale Hypertension eine deutliche Prognoseeinschränkung bedeuten.12
Literatur:
1 European Association for the Study of the Liver (electronic address: easloffice@easloffice.eu ): EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol 2017; 67(1): 145-72 2 Corpechot C et al.: Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology 2008; 48(3): 871-73 Harms MH et al.: Major hepatic complications in ursodeoxycholic acid-treated patients with primary biliary cholangitis: risk factors and time trends in incidence and outcome. Am J Gastroenterol 2018; 113(2): 254-64 4 Nevens F et al.: A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016; 375(7): 631-43 5 Corpechot C et al.: A placebo-controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med 2018; 378(23): 2171-81 6 Tanaka A et al.: Association of bezafibrate with transplant-free survival in patients with primary biliary cholangitis. J Hepatol 2021; 75(3): 565-71 7 Burghart L et al.: Distinct prognostic value of different portal hypertension-associated features in patients with primary biliary cholangitis. J Gastroenterol 2021; 57(2): 99-110 8 European Association for the Study of the Liver: EASL Clinical Practice Guidelines: Autoimmune hepatitis. J Hepatol 2015; 63(4): 971-1004 9 Komori A: Recent updates on the management of autoimmune hepatitis. Clin Mol Hepatol 2021; 27(1): 58-69 10 Hennes EM et al.: Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008; 48(1): 169-76 11 Boonstra K et al.: Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013; 58(6): 2045-55 12 Rabiee A, Silveira MG: Primary sclerosing cholangitis.Transl Gastroenterol Hepatol 2021; 6: 29 13 Weismüller TJ et al.: Patient age, sex, and inflammatory bowel disease phenotype associate with course of primary sclerosing cholangitis. Gastroenterology 2017; 152(8): 1975-84.e8 14 de Vries EMG et al.: Alkaline phosphatase at diagnosis of primary sclerosing cholangitis and 1 year later: evaluation of prognostic value. Liver Int 2016; 36(12): 1867-75 15 Kaplan GG et al.: The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis. Am J Gastroenterol 2007; 102(5): 1042-9 16 Boberg KM et al.: Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol 2011; 54(2): 374-85 17 Burak KW et al.: Is there a role for liver biopsy in primary sclerosing cholangitis? Am J Gastroenterol 2003; 98(5): 1155-8 18 Chapman R et al.: Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010; 51(2): 660-78 19 European Association for the Study of the Liver: EASL Clinical Practice Guidelines: Management of cholestatic liver diseases. J Hepatol 2009; 51(2): 237-67 20 Shi J et al.: Ursodeoxycholic acid in primary sclerosing cholangitis: meta-analysis of randomized controlled trials. Hepatol Res 2009; 39(9): 865-73 21 Lazaridis KN, LaRusso NF: Primary sclerosing cholangitis. N Engl J Med 2016; 375(25): 2501-2 22 Bambha K et al.: Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology 2003; 125(5): 1364-9
Das könnte Sie auch interessieren:
UEGW 2025: Neues aus der Hepatologie
In der Indikation metabolisch bedingte Lebersteatose bzw. Steatohepatitis scheiterten über viele Jahre zahlreiche Therapieversuche. Seit Kurzem stehen erstmals wirksame Therapien zur ...
Fünf Therapien zur Behandlung von MASLD
Mit zunehmendem Übergewicht in der Bevölkerung wird auch die mit metabolischer Dysfunktion assoziierte steatotische Lebererkrankung (MASLD) vermehrt diagnostiziert. PD Dr. Dr. med. David ...
Sonografie der Leber – aktuelle Entwicklungen, quantitative Verfahren und klinische Bedeutung
Der vorliegende Übersichtsartikel fasst aktuelle technische Entwicklungen sowie klinische Anwendungen und Limitationen der Lebersonografie zusammen und diskutiert deren Stellenwert im ...