Syndromes myélodysplasiques/néoplasies myélodysplasiques, où va-t-on?
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Deux classifications actualisées des lymphomes et des néoplasies myéloïdes couvrent des domaines thématiques similaires, mais diffèrent sur certains points pertinents.
Dans notre monde complexe, on constate de manière perceptible et objective une augmentation de la polarisation.1 Celle-ci n’a pas épargné l’Academia. Quelque 28ans seulement après les efforts internationaux exemplaires pour éliminer toutes les différences dans les classifications des tumeurs hématolymphoïdes, qui ont abouti à la création de la troisième, quatrième et quatrième édition révisée de la classification OMS, toutes universellement reconnues, les forces centrifuges ont pris le dessus dans la communauté des hématopathologues et des hémato-oncologues dans les années 2021–2022.
Il en a résulté l’élaboration de deux classifications indépendantes à la fois des lymphomes et des néoplasies myéloïdes, qui ont été publiées peu de temps après, à l’été 2022, dans les revues renommées Blood2 et Leukemia3: les classifications ICC2022 et OMS5.
Perdu au fil de la traduction: SMD
Même si les deux classifications se recoupent largement (Tab.1)4, néanmoins, le syndrome myélodysplasique (SMD) est mentionné dans les classifications sous deux noms différents, à savoir «syndrome myélodysplasique» (SMD) dans la classification ICC20222 et «néoplasie myélodysplasique» dans la classification OMS5, qui est toujours traditionnellement abrégée SMD (plutôt que NMD).3 De plus, certaines sous-entités ne sont pas mentionnées dans une classification et pas l’autre, et les critères de classification de certaines sous-entités présentent des différences notables.
Si l’on étudie les deux énoncés de position sur les classifications2,3, on constate que si les preuves scientifiques pour la nouvelle édition de la classification OMS4 révisée, sur laquelle sont basées les deux classifications, ICC2022 et OMS5, sont quasiment identiques, elles ont été interprétées différemment.
En raison de la subjectivité potentielle, l’ICC accorde moins d’importance à la recherche de dysplasie (Fig. 1A et B) et à la quantité exacte de blastes5, et met davantage l’accent sur les anomalies génétiques récurrentes sous-jacentes. La classification OMS suit davantage le modèle de classification traditionnel du SMD.6
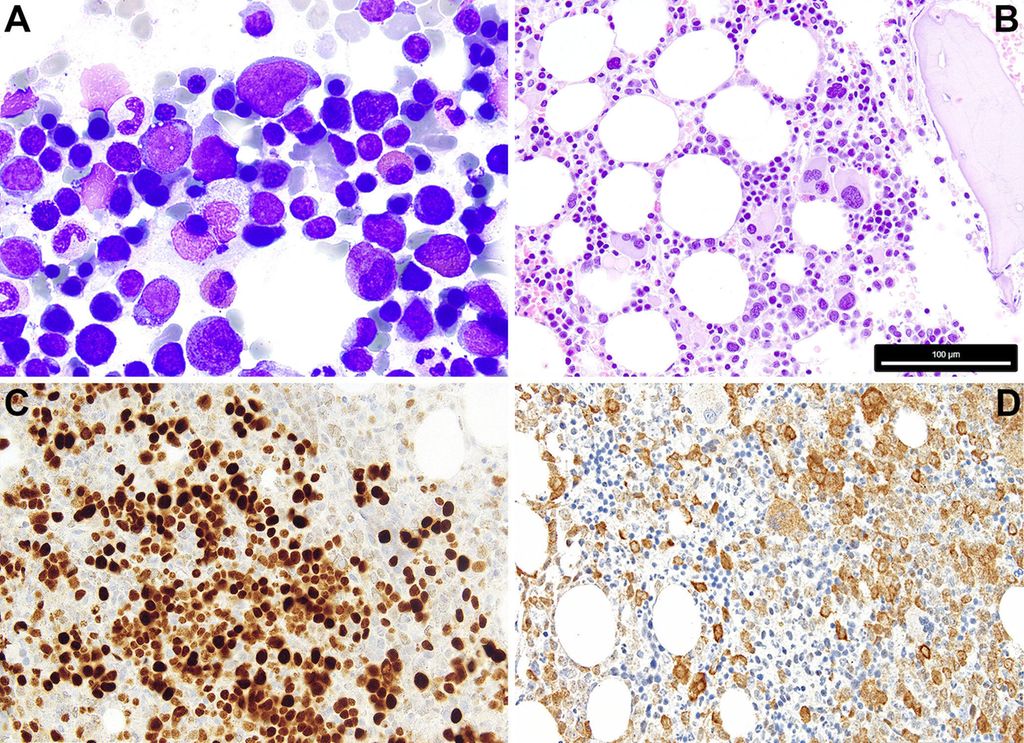
Fig. 1: 1A: Cytologie d’un SMD avec excès de blastes/blastes multipliés; on remarque les petits mégacaryocytes dysplasiques, le trouble de la maturation et les défauts de granulation de la myélopoïèse, la dyserythropoïèse clairsemée et 7% de blastes. 1B: Histopathologie d’un SMD avec del(5q) et mutation monoallélique TP53; on note la présence de quelques mégacaryocytes monolobés nucléaires de petite taille, à côté de mégacaryocytes à noyaux séparés, et le trouble de la maturation myélopoïétique. 1C: Positivité caractéristique de p53 à l’immunohistochimie dans le SMD avec mutations TP53 multi-hit/inactivation biallélique de TP53 (6% de blastes); la protéine p53 mutée dispose d’une demi-vie plus longue et stabilise l’éventuelle p53 de type sauvage (encore) présente, de sorte que la protéine correspondante, qui n’est sinon guère détectable à l’immunohistochimie dans la moelle osseuse, peut être colorée en cas de charge allélique mutée significative, comme dans le cas de mutations TP53 multi-hit/inactivation TP53biallélique. 1D: SMD avec excès de blastes (8% de blastes) et positivité avec l’anticorps anti-NPM1 spécifique de la mutation (PA1-46356); selon l’OMS, le cas doit être classé comme LMA, alors que selon l’ICC, un diagnostic de SMD avec excès de blastes est admissible. La barre de mesure dans B se réfère aux Fig. B–D
Cela se reflète également dans la définition du SMD: Si la présence d’au moins une cytopénie inexplicable (à savoir anémie avec hémoglobine <120g/l pour les femmes et <130g/l pour les hommes, neutropénie <1,8x109/l et/ou thrombopénie <150x109/l) est présente dans les deux classifications, la présence d’anomalies génétiques définissant le SMD – del(5q), mutations TP53 multi-hit, mutations SF3B1, -7/del(7q) ou caryotype complexe – la présence de dysplasie n’est plus exigée dans la classification ICC pour le diagnostic et les cas correspondants sont classés comme «SMDsans dysplasiemorphologique». En revanche, à l’exception du «SMDavec faible quantité de blastes» et de la «délétion 5qisolée», l’OMS classe les éventuels cas sans dysplasie comme des cytopénies clonales de signification indéterminée (CCSI).
À cet égard, l’ICC a accordé un poids diagnostique à la forte signification pronostique, en l’occurrence positive, et aussi prédictive de del(5q) (relativement à la sensibilité au lénalidomide), mais surtout à la signification pronostique négative des autres anomalies génétiques énumérées7,8, alors que l’OMS ne le prévoit que pour del(5q). Cela donne également lieu à des difficultés de traduction (Tab. 1).
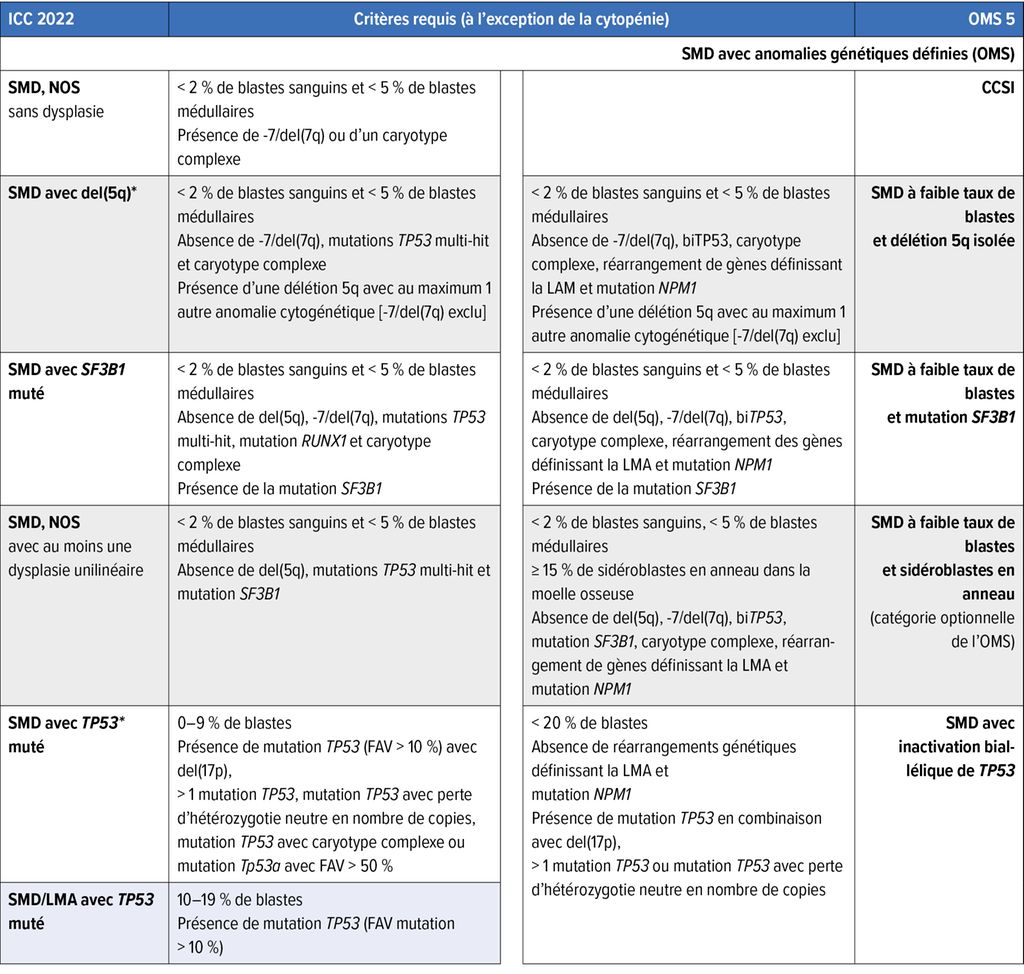
Tab. 1: Comparaison de la classification des syndromes myélodysplasiques (SMD)/néoplasies myélodysplasiques: International Consensus Classification 2022 (ICC 2022) vs 5e classification de l’Organisation mondiale de la Santé (OMS5); les catégories sur fond coloré ne figurent pas dans l’autre classification
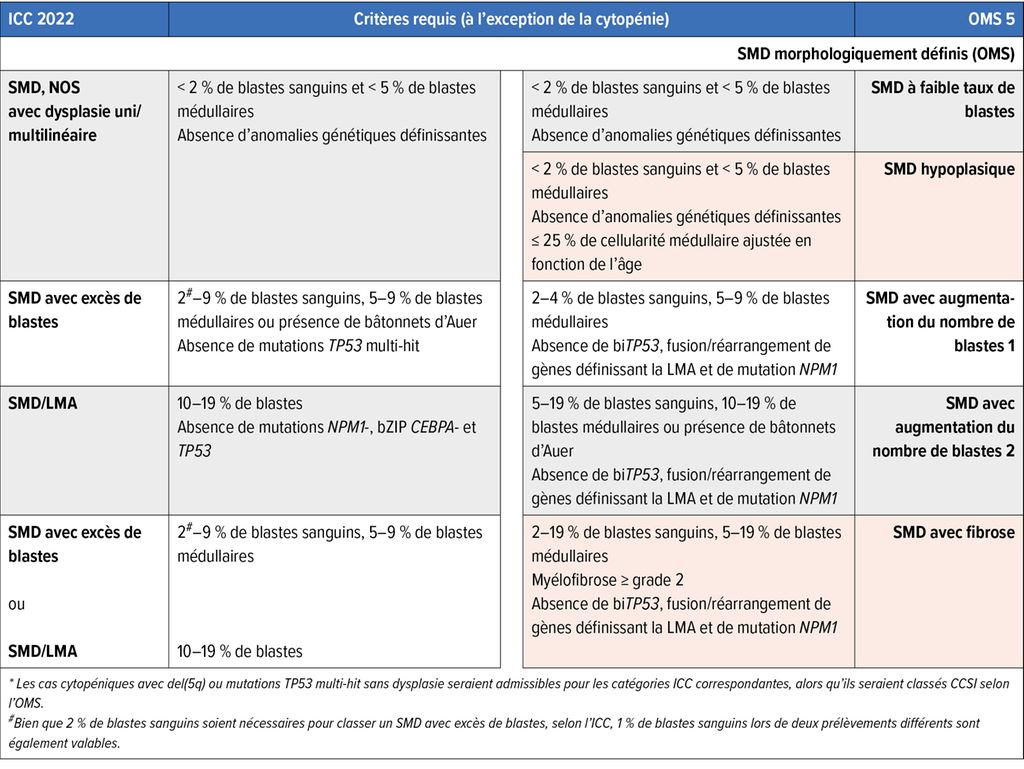
Tab.1: (suite)
Toutefois, à cette exception près, les deux classifications exigent l’identification de >10% de dysplasies dans les séries de maturation hématopoïétique pour distinguer le SMD du CCSI dans tous les autres cas.
Difficultés de délimitation: LMA
Le cas des leucémies myéloïdes aiguës (LMA) poste d’autres difficultés de délimitation. Ici, la présence d’anomalies génétiques définissant la LMA entraîne, à quelques exceptions près et indépendamment de la quantité de blastes, le diagnostic de l’OMS de LMA3. Selon la classification ICC2, ce n’est le cas qu’à partir d’une quantité de blastes >10%. Par exemple, un SMD avec une mutation NPM1 n’est pas un diagnostic acceptable selon l’OMS, alors que c’est acceptable selon les critères de l’ICC jusqu’à une quantité de blastes <10% (Fig.1D).
C’est précisément dans la délimitation entre LMA et SMD qu’apparaît une autre différence pertinente entre les deux classifications: l’ICC a déclaré l’entité «SMD/LMA» pour les cas présentant 10–19% de blastes dans le sang périphérique et/ou la moelle osseuse en l’absence d’anomalies génétiques définissant une LMA (ne s’applique pas aux SMD pédiatriques [<18ans]).2 La création de cette catégorie est étayée par le fait que le pronostic des patient·es atteint·es de LMA oligoblastique (20–30% de blastes) est comparable à celui des patient·es de la catégorie de l’OMS du «SMD avec augmentation du nombre de blastes2» (qui n’est plus répertoriée par l’ICC).5 L’avantage pratique est que la nouvelle catégorie SMD/LMA formalise l’inclusion des patient·es dans les études sur le SMD et la LMA et peut ainsi faciliter les négociations avec les organismes payeurs, ce qui, il est vrai, est également abordé par l’OMS3, mais n’est pas inclus dans la classification.
Abandon des sidéroblastes en anneau?
La catégorie de l’ICC «SMDavec SF3B1 muté»/catégorie de l’OMS «SMD avec faible quantité de blastes et SF3B1 muté» remplace la plupart des cas de l’ancienne catégorie de l’OMS4 «SMDavec sidéroblastes en anneau» (SMD-SA). La nouvelle classification ICC renonce complètement aux SMD-SA: les cas de sidéroblastes en anneau en l’absence de mutation SF3B1 sont classés comme «SMD, NOS», alors que l’OMS les classe de manière optionnelle comme «SMD avec faible quantité de blasteset sidéroblastes en anneau» (si ceux-ci représentent ≥15%).
La justification de la classification ICC est que la biologie et le pronostic favorable des cas de SMD avec sidéroblastes en anneau et mutation SF3B1 sont déterminés par la présence de la mutation [lorsque – mentionné explicitement par l’ICC2 – elle n’est pas accompagnée de del(5q), d’un caryotype défavorable, de mutations RUNX1 ou TP53] et non par la présence de sidéroblastes en anneau.9 Néanmoins, l’ICC conseille également de rechercher activement les sidéroblastes en anneau, car ils sont l’expression d’une dysérythropoïèse et contribuent donc au diagnostic de SMD, et leur présence prédit une éventuelle mutation SF3B1 et, éventuellement, une réponse au luspatercept10.
Mutations TP53
Les deux classifications accordent une grande importance aux mutations TP538, 11 et introduisent désormais les catégories de «SMD avec mutations TP53 multi-hit » (0–9% de blastes) et de «SMD/LMAavec TP53 muté» (10–19% de blastes; ICC) ou de «SMD avec inactivation TP53 biallélique» (biTP53; 0–19% de blastes; OMS), qui l’emportent sur toutes les autres catégories de SMD. Multi-hit ou biTP53 est défini par une mutation TP53 combinée à del(17p), >1 mutation TP53 ou une mutation TP53 avec perte d’hétérozygotie neutre en nombres de copies2,3, ce qui se traduit souvent par une fréquence allélique des variants (FAV) >50%. À cet égard, l’ICC admet également les mutations TP53 conjointement à un caryotype plus complexe et exige explicitement une FAV ≥10% pour les mutations TP53.2
Une différence très importante entre les deux classifications apparaît dans les cas avec 10–19% de blastes: dans de tels cas, selon l’ICC, comme le suggèrent également les données des publications11, la présence d’une mutation TP53 suffit pour poser le diagnostic de SMD/LMA avec TP53 muté2, alors que pour l’OMS, un statut biTP53 doit être présent pour poser le diagnostic de SMD-biTP533.
Lacune dans la classification ICC: SMD hypoplasique
Le SMD hypoplasique (SMD-h) est exclusivement répertorié par l’OMS et est défini comme un SMD sans anomalies génétiques définissantes avec <5% de blastes et ≤25% de cellularité de la moelle osseuse ajustée selon l’âge.3 Le SMD-h est connu pour ses caractéristiques12, notamment sa réponse potentielle à l’immunosuppression13, et semble également présenter un profil spécifique avec moins de mutations génétiques codant pour le facteur d’épissage et plus de mutations codant pour les hélicases à ARN14, de sorte que le fait qu’il ne soit pas répertorié par l’ICC constitue une lacune.
Cela ne semble pas être le cas pour la deuxième catégorie, qui n’est pas répertoriée par l’ICC, à savoir le «SMD avec fibrose»3: dans le cas du SMD, même si la fibrose est un paramètre pronostique indépendant du nombre de blastes15, elle est étroitement liée à l’âge, au nombre de blastes, à la catégorie SMD de l’OMS4, aux mutations TP53 et SETBP1 et à un caryotype de mauvais pronostic16, et un seuil unique, le degré de fibrose ≥2 ou ≥315,16, n’est pas universellement reconnu, de sorte qu’il n’est pas clair si le degré de myélofibrose ≥2 dans les cas du SMD avec 5–19% de blastes médullaires est l’expression d’une biologie spécifique, comme le suggère l’OMS3, ou représente un épiphénomène mémorable.
Conclusion
Le degré de complexité du diagnostic de SMD a augmenté avec l’introduction des deux classifications en 2022. Outre les informations cliniques minimales requises (hémogramme différentiel, anamnèse pour le traitement cytotoxique, anamnèse familiale pour prédisposition à la lignée germinale), les résultats des tests de laboratoire (dyshématopoïèse et quantité de blastes médullaires, caryotypage pour les anomalies spécifiques du SMD ou pour les anomalies de la lignée germinale) sont indispensables pour exclure les anomalies spécifiques à la LMA et les tests de mutation, au moins pour SF3B1, TP5317, NPM118 et CEBPA; Fig.1C et D), ainsi qu’une traduction des sous-catégories spécifiques de SMD, du libellé de l’ICC à celui de l’OMS et vice versa.
Littérature:
1 Ross Arguedas A et al.: Oxford 2022: Reuters Institute, University of Oxford. En ligne sous https://reutersinstitute.politics.ox.ac.uk/echo-chambers-filter-bubbles-and-polarisation-literature-review . Consulté le 29décembre 2023 2Arber DA et al.: Blood 2022; 140(11): 1200-28 3 Khoury JD et al.: Leukemia 2022; 36(7): 1703-19 4 Zeidan AM et al.: Leukemia 2022; 36(12): 2939-46 5 Estey E et al.: Blood 2022; 139(3): 323-32 6 Bennett JM et al.: Br J Haematol 1982; 51(2): 189-99 7 Bernard E et al.: Nat Med 2020; 26(10): 1549-56 8 Grob T et al.: Blood 2022; 139(15): 2347-54 9 Malcovati L et al.: Blood 2020; 136(2): 157-70 10 Amitai I et al.: Blood 2023; 142(Suppl. 1): 6487 11 Weinberg OK et al.: Blood Adv 2022; 6(9): 2847-53 12 Bono E et al.: Leukemia 2019; 33(10): 2495-505 13 Passweg JR et al.: J Clin Oncol 2011; 29(3): 303-9 14 Nazha A et al.: Haematologica 2015; 100(11): e434-7 15 Della Porta MG et al.: J Clin Oncol 2009; 27(5): 754-62 16 Melody M et al.: Clin Lymphoma Myeloma Leuk 2020; 20(5): 324-8 17 McGraw KL et al.: Haematologica 2016; 101(8): e320-3 18 Patel SS et al.: Mod Pathol 2020; 33(7): 1380-8
Das könnte Sie auch interessieren:

Cancer de l’endomètre métastatique, avancé et récidivant
L’association d’inhibiteurs de point de contrôle et de chimiothérapie peut être considérée comme une nouvelle norme thérapeutique dans les cancers de l’endomètre avec déficit de ...

Parfois, il suffit d’attendre
Compte tenu de la multitude d’options thérapeutiques ciblées et immunothérapeutiques désormais disponibles, la prise en charge du carcinome à cellules rénales en situation adjuvante et ...

Les néoplasies myélodysplasiques: stratégies thérapeutiques personnalisées
Les progrès réalisés dans la compréhension moléculaire des néoplasies myélodysplasiques (MDS, myelodysplastic syndrome) ont permis de mettre au point des approches thérapeutiques ...