
Epidermolysis bullosa: Hürden in der Etablierung neuer Therapien
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Das komplexe und diverse Krankheitsbild der Epidermolysis bullosa macht es erforderlich, Patient:innen individuell zu beurteilen und zu therapieren. Neue Erkenntnisse in Bezug auf die zugrunde liegenden systemischen Entzündungsreaktionen haben in den letzten Jahren zur Entwicklung neuer Therapieansätze geführt. Diese lassen auf effektive Behandlungsmöglichkeiten in der Zukunft hoffen, bringen aber auch Herausforderungen mit sich.
Keypoints
-
Die Epidermolysis bullosa (EB) bezeichnet eine heterogene Gruppe erblich bedingter Krankeiten, die mit massiv gesteigerter Hautsensibilität, Blasenbildung und chronischen Wunden einhergeht.
-
Neue Erkenntnisse zur entzündlichen Komponente der EB haben in den letzten Jahren zur Entwicklung neuer Therapieansätze geführt, die bei der Entzündungsmodulation ansetzen.
-
Die neuen Therapieoptionen fokussieren auf palliative bzw. krankheitsmodifizierende, aber auch potentiell kurative Behandlungsstrategien. Seit Kurzem gibt es erstmals spezifische topische Medikamente zur Behandlung der EB.
-
Die geringe Anzahl von Patienten, die für klinische Studien infrage kommen, sowie die Heterogenität der Krankheit stellen große methodische Herausforderungen an die Foschung.
Epidermolysis bullosa (EB) bezeichnet eine erbliche, heterogene Gruppe seltener Erkrankungen, die mit gesteigerter, mechanisch präzipitierter mukokutaner Fragilität, Blasenbildung und chronischen Wunden einhergeht. EB beruht auf Mutationen in zumindest 16 verschiedenen Genen, die überwiegend für Struktur- und Adhäsionsproteine der dermoepidermalen Junktionszone (DEJ) codieren und deren strukturelle und funktionelle Integrität aufrechterhalten. Eine zusätzliche Expression der Indexgene in anderen epithelialen Geweben und mesenchymalen Organen begründet das Auftreten von primär extrakutanen EB-Manifestationen an Schleimhäuten und inneren Organen.
Das überaus breite phänotypische Spektrum von EB mit potenziell schweren, extrakutanen Symptomen ist neben der genetischen Heterogenität auf den Einfluss u.a. epigenetischer, umweltbezogener, biochemischer sowie sozioökonomischer Faktoren zurückzuführen. So werden mehr als 30 EB-Subtypen unterschieden, deren Klassifikation auf verschiedenen Phänotypen, Vererbungsmustern, Genen und Mutationen basiert und die subtypspezifische Unterschiede hinsichtlich Ausdehnung, Verteilung, Schweregrad und potenzieller Komplikationen aufweisen. EB wird in 4 Haupttypen in Bezug auf die Ebene der Spaltung innerhalb der Haut eingeteilt: EB simplex (EBS) mit epidermaler Spaltbildung, junktionale EB (JEB) mit Spaltbildung innerhalb der dermoepidermalen Junktionszone, dystrophe EB (DEB) mit dermaler Spaltbildung und Kindler-EB (KEB) mit Spaltbildung in unterschiedlichen Ebenen der DEJ.1
Der häufigste Typ, EBS, präsentiert sich mit prominenter Blasenbildung, während bei JEB chronische, teils granulierende Wunden im Vordergrund stehen. Als weitere Symptome sind Alopezie und die Mitbeteiligung von Organen (z.B. Niere, ableitende Harnwege) häufig. Bei DEB treten neben chronischen und rekurrenten Wunden auch Nageldystrophie und -verlust auf. Ausgeprägt ist die Narbenbildung, die zu Pseudosyndaktylie (fibrotisch bedingte Verwachsungen von Fingern, Zehen) führt. Auf dem Boden chronischer Traumatisierung und gestörter Wundheilung treten bereits im jungen Erwachsenenalter Plattenepithelkarzinome auf, die aufgrund ihres aggressiven Verlaufes eine deutlich eingeschränkte Lebenserwartung bedingen. Die Hauptmerkmale von KEB sind mukokutane Erosionen, atrophe Narben, Lichtempfindlichkeit sowie Poikilodermie und Parodontalerkrankungen. Schwere EB-Formen (insbesondere JEB und DEB) werden von extrakutanen Manifestationen und Komplikationen begleitet, die u.a. Dystrophie und Wachstumsverzögerung, Anämie, Infektionen inklusive Sepsis, exzessive Karies, Ösophagusstrikturen, Atemwegsstenosen, therapierefraktären Juckreiz und ausgeprägte Schmerzen, aber auch renale Amyloidose oder Kardiomyopathie umfassen können.2,3
Das vielfältige Symptomenspektrum verursacht eine erhebliche Beeinträchtigung der Lebensqualität Betroffener und unterstreicht die dringende Notwendigkeit effektiver Therapien für diese chronische, zeitlebens bestehende und häufig progressive Erkrankung.
Ein rezenter systematischer Review zur Krankheitslast bei RDEB betont die signifikanten negativen ökonomischen, sozialen und klinischen Auswirkungen der Erkrankung. Dabei zeigt sich beispielsweise, dass bei 60% der Patienten chronische Wunden, die mehr als 30% der Körperoberfläche umfassen, vorhanden sind und die Wundgröße positiv mit Chronizität, Schmerzen und Juckreiz korreliert. Plattenepithelkarzinome traten in der Kohorte bei 67% der Patienten auf und waren mit einer Mortalität von 84% bis zum 40. Lebensjahr vergesellschaftet. 80% der Familien Betroffener berichteten über negative Auswirkungen der Erkrankung auf ihr persönliches Leben. Über 50% litten unter der finanziellen Belastung durch die Erkrankung. Die regelmäßig notwendigen Verbandwechsel verursachten einen erheblichen Zeitaufwand für Betroffene (15–40% der Patienten benötigten 3 Stunden für einen Verbandwechsel), der die Gestaltung des Alltages maßgeblich prägt.4
Chronische systemische Entzündungsreaktionen bei schweren Formen von EB
Die chronische Gewebetraumatisierung im Rahmen der EB induziert und unterhält eine im Verlauf unproduktive Entzündungsreaktion, die den kumulativen Gewebeschaden lokal, aber auch mit systemischer Wirkung, verstärkt. Erhöhte proinflammatorische Zytokine wie IL-1β, IL-6, „transforming growth factor β“ (TGF-β) und Interferon γ wurden bei EB dementsprechend in Gewebe, Blasenflüssigkeiten und auch im Serum nachgewiesen. Insbesondere schwere EB-Formen mit generalisiertem Haut- und Schleimhautbefall stellen daher in Analogie zur Psoriasis oder atopischen Dermatitis eine systemisch wirkende entzündliche Erkrankung dar.5–8 Ausgehend von einer Barrierestörung, kommt es demnach durch transkutane Invasion von Reizstoffen, Mikroben und Allergenen zur Induktion von pro- und autoinflammatorischen Signalwegen, die Dysbiose, Erschöpfung des Hautstammzellenpools und prokanzerogene Umbauprozesse im Bindegewebe unterhalten.8
Diese Erkenntnisse zur entzündlichen Krankheitskomponente der EB führten in den letzten Jahren zur Entwicklung neuer Therapieansätze, die sich gegen die bei EB aktiven Entzündungsmediatoren und -kaskaden richten (z.B. IL-1β-Inhibition mit Diacerein bei EBS).
Zulassung neuer Therapieoptionen für seltene Erkrankungen
Auf translationaler Ebene haben mehrere therapeutische Ansätze für EB den Weg in die klinische Erprobung gefunden. Dabei stehen neben palliativen (z.B. juckreizhemmenden) bzw. krankheitsmodifizierenden (z.B. immunmodulatorischen) Behandlungsstrategien auch potenziell kurative Therapiemodalitäten (Gentherapie) im Vordergrund (Abb. 1).9
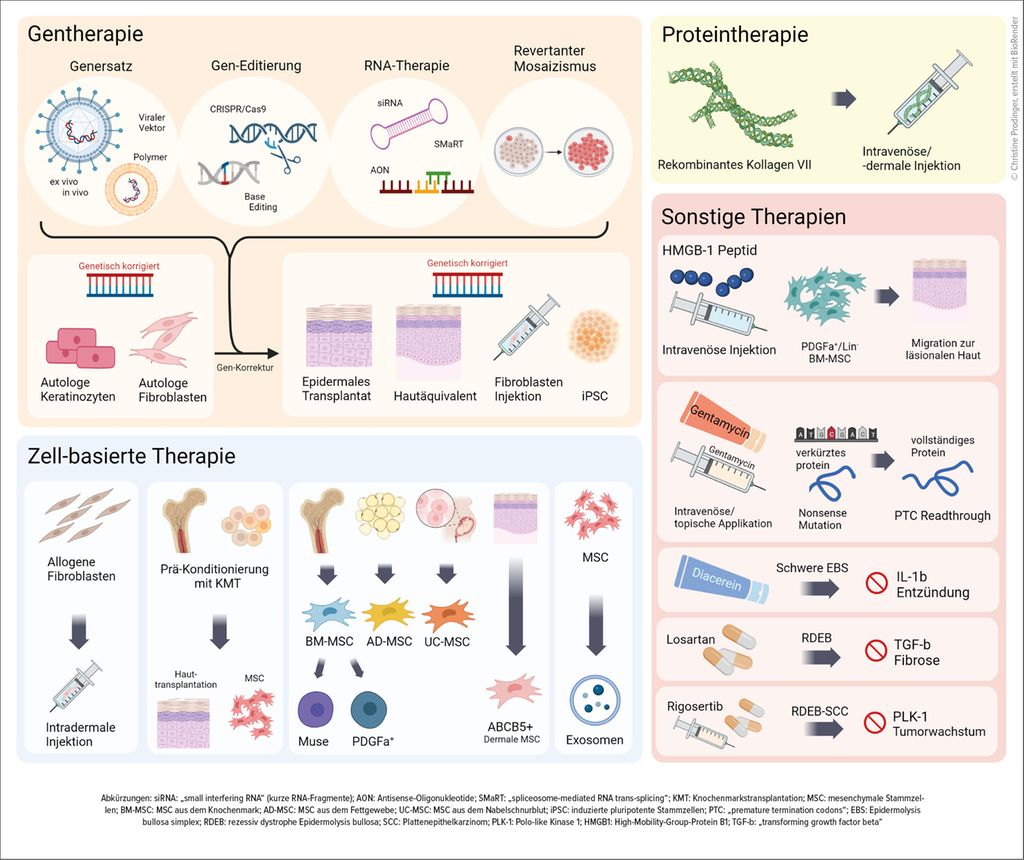
Abb. 1: Investigative Therapien für Epidermolysis bullosa (Auswahl)9
Das erste spezifisch für bestimmte Formen der EB zugelassene topische Medikament kam im Juni 2022 in Europa auf den Markt. Filsuvez®-Gel enthält trockenen Birkenrindenextrakt, der entzündungs- und juckreizhemmend sowie wundheilungsfördernd wirken soll. Im Mai 2023 wurde von der FDA zudem eine topische Genersatztherapie (Vyjuvek®) für DEB-Patienten ab dem 6. Lebensmonat mit Mutationen im COL7A1-Gen zugelassen, wo ein replikationsdefekter HSV-1-Vektor das Korrekturgen (COL7A1-Gen) episomal in die Hautzellen offener Wunden einbringt und so die Wundheilung signifikant verbessert.
Gemeinsame pathogenetische Endstrecken bzw. Kollateralschäden bilden die Grundlage für Drug-Repurposing-Ansätze bei EB. Dabei werden mehrere bereits am Markt erhältliche Medikamente unmittelbar bei EB eingesetzt bzw. in Studien untersucht (unter anderem Losartan, Dupilumab, Diacerein, Calcipotriol, Gentamycin, JAK-Inhibitoren, PD-1-Inhibitoren).10
Moderne molekularbiologische Techniken und Hochdurchsatzverfahren ermöglichen immer effizienter und auch individueller, Schlüsselstrukturen und/oder Mediatoren als therapeutische Ziele für spezifische Interventionen zu identifizieren. So konnten mit Transkriptom-Analysen von Material von 11 EBS-Patienten mehr als 1200 im Vergleich zu Kontrollproben dysregulierte Gene und eine signifikante Hochregulierung der IL-6-, IL-8- und IL-10-Signalwege nachgewiesen werden. Die Korrelation dieses molekularen Profils mit Signaturen aus Zelllinien, die mit 2423 Medikamenten behandelt wurden, führte zur Identifikation von mTOR-Inhibitoren und Phosphatidylinositol-3-Kinase-Inhibitoren als vielversprechendste Kandidaten, die dem pathologischen Profil am besten entgegenwirkten. Eine im Anschluss durchgeführte Pilotstudie mit topischem Sirolimus bei zwei EBS-Patienten wies eine deutliche klinische Verbesserung nach.11
Herausforderungen neuer Therapieoptionen am Beispiel Rigosertib
Rigosertib ist ein Polo-like-Kinase(PLK)-Inhibitor, der mit verschiedenen molekularen Signalwegen interferiert, die für das Tumorwachstum entscheidend sind. Präklinische Daten zeigten hohe Effektivität und Selektivität in vitro und in vivo gegen RDEB-Tumor-Keratinozyten bei geringer Toxizität.12 Eine 24-jährige RDEB-Patientin mit Zustand nach mehreren rezidivierenden und zuletzt inoperablen Plattenepithelkarzinomen wurde nach fehlendem Ansprechen auf Cemiplimab (PD-1-Antikörper) sodann als erste Patientin weltweit in eine Studie des EB-Haus Austria aufgenommen und es wurde ihr Rigosertib verabreicht.
Die Patientin erhielt die Therapie intravenös (IV) (8x 2-Wochen-Zyklus, dann 4-Wochen-Zyklus) und zeigte bereits innerhalb von 9 Wochen nach Einleitung ein sehr gutes Ansprechen mit Komplettregression der an den Extremitäten vorhandenen Plattenepithelkarzinome. Die Tumorremission bestand über die gesamte 2-jährige Behandlungsperiode. Allerdings zeigten sich insbesondere im ersten Jahr erhebliche Hindernisse bei der Applikation der Medikation. Eine orale Einnahme von Rigosertib ist für die meisten RDEB-Patienten aufgrund der Größe der Kapsel und häufigen Ösophagusstenosen nicht möglich. Die intravenöse Verabreichung, welche protokollgemäß über einen Zeitraum von 72 Stunden als kontinuierliche IV-Infusion alle 2 bzw. 4 Wochen zu erfolgen hat, ist nicht nur zeitaufwendig, sondern birgt ob der großflächigen Hautwunden auch ein signifikantes Infektionsrisiko für die Patienten. Dementsprechend traten bei der Patientin trotz antibiotischer Prophylaxe auch wiederholt Bakteriämien und septische Zustandsbilder durch Port-a-Kath-Infektionen und PICC-Katheter-Anlagen auf, was den Krankenhausaufenthalt der Patientin signifikant verlängerte. Zudem bereitete die EB-inhärente Hämaturie anfänglich differenzialdiagnostische Schwierigkeiten in der Abgrenzung einer medikamentös induzierten toxisch-irritativen Cystitis. Letztere wurde schließlich verifiziert und veranlasste uns zu einer Dosisreduktion von Rigosertib auf 50%.
Herausforderung klinischer Studien bei seltenen Erkrankungen
Klinische Studien sind essenziell für die Evaluierung neuer Therapieoptionen. Die geringe Anzahl von (eligiblen) Patienten und die Heterogenität der Krankheit wie auch der Patientenkohorte stellen große methodische Herausforderungen für die Generierung von direktiven Studiendaten dar (z.B. Rekrutierungspotenzial, -dauer; Studiendauer und -kosten; Signifikanzniveau der analysierbaren Daten). Um dem zu begegnen, werden häufig komplexe Studiendesigns entwickelt, die mit einer beträchtlichen Belastung der Teilnehmenden durch beispielsweise häufige Visiten/Messpunkte mit wiederholt langer und damit beschwerlicher Anreise zum Studienzentrum oder auch durch kritisch-invasive Diagnostiken wie Blutabnahmen und Hautbiopsien einhergehen. Das hat rückwirkend wiederum negative Auswirkungen auf die Rekrutierung.
Vor diesem Hintergrund ist eine patientenzentrierte Herangehensweise, die die Bedürfnisse der Betroffenen berücksichtigt, entscheidend für die erfolgreiche Durchführung klinischer Studien. Dabei verbessern eine intensive Planung und Zusammenarbeit von Sponsoren, Prüfärzten, Forschern, Gesundheitsbehörden und Patientengruppen eine effiziente Umsetzung, was in der Folge den Zugang zu wirksamen Medikamenten beschleunigt.13
Begrenzt verfügbare prospektive und longitudinale Daten zur EB als seltene Erkrankung erschweren zudem die Bestimmung wichtiger studienprotokollbezogener Parameter wie Meilensteine, geeignete Effektgrößen und Endpunkte. Ein kürzlich durchgeführter Scoping Review zu klinischen Studien bei EB ergab, dass alleine für diese Erkrankung insgesamt 1280 verschiedene Endpunkte und 200 Messinstrumente Verwendung im Rahmen klinischer Prüfungen fanden.14 Diese ausgeprägte Heterogenität erschwert die Vergleichbarkeit klinischer Studien, die Integration von Daten zu einer seltenen Erkrankung und damit die ehestmögliche Verfügbarkeit der effizientesten Therapien.
Innovative Technologien wie Big Data Management, Omics, Digitalisierung, Bildverarbeitungssysteme, Automatisierung und KI sollen (zukünftig) helfen, die vielschichtige Komplexität der Erkrankung EB und ihrer qualitätsgesicherten Versorgung besser zu adressieren, u.a. durch die Generierung quantitativ und qualitativ optimierter klinischer Information (Patientenidentifikation, Biomarker, Modellsysteme) oder effizienteres Drug Discovery und Drug Development. Neben Patientenzentrizität sind dazu bei EB ein global-kollaborativer Ansatz und eine Harmonisierung regulatorischer Anforderungen vonnöten. Es bleibt zu hoffen, dass die Vielzahl an innovativen Entwicklungen und die wahrnehmbare Dynamik alsbald den Patienten erlebbar zugutekommen werden.
Literatur:
1 Has C et al.: Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol 2020; 183(4): 614-27 2 Fine JD, Mellerio JE: Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol 2009; 61(3): 367-84 3 Fide JD, Mellerio JE: Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol 2009; 61(3): 387-402 4 Tang JY et al.: A systematic literature review of the disease burden in patients with recessive dystrophic epidermolysis bullosa. Orphanet J Rare Dis 2021; 16(1): 175 5 Breitenbach JS et al.: Transcriptome and ultrastructural changes in dystrophic epidermolysis bullosa resemble skin aging. Aging (Albany NY) 2015; 7(6): 389-411 6 Annicchiarico G et al.: Proinflammatory cytokines and antiskin autoantibodies in patients with inherited epidermolysis bullosa. Medicine (Baltimore) 2015; 94(42): e1528 7 Haghighi Javid A et al.: Interleukin-17A immune pattern across genetic acantholytic and blistering disorders. Clin Exp Dermatol 2023; 48(5): 518-23 8 Prodinger C et al.: Translational perspectives to treat epidermolysis bullosa-Where do we stand? Exp Dermatol 2020; 19(11): 1112-22 9 Hou PC et al.: Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol 2021; 22(6): 801-17 10 Zauner R et al.: Transcriptome-guided drug repurposing for aggressive SCCs. Int J Mol Sci 2022; 23(2): 1007 11 Lee GH et al.: Transcriptomic repositioning analysis identifies mTOR inhibitor as potential therapy for epidermolysis bullosa simplex. J Invest Dermatol 2022; 142(2): 382-9 12 Atanasova VS et al.: Identification of rigosertib for the treatment of recessive dystrophic epidermolysis bullosa-associated squamous cell carcinoma. Clin Cancer Res 2019; 25(11): 3384-91 13 Prodinger C et al.: Profiling trial burden and patients’ attitudes to improve clinical research in epidermolysis bullosa. Orphanet J Rare Dis 2020; 15(1): 182 14 Korte EWH et al.: Heterogeneity of reported outcomes in epidermolysis bullosa clinical research: a scoping review as a first step towards outcome harmonization. Br J Dermatol 2023; 189(1): 80-90
Das könnte Sie auch interessieren:

Aminosäuren – Booster für die Wundheilung?
Für den Wundheilungsprozess ist je nach Heilungsprozess die richtige Kombination aus Kohlenhydraten, Fetten und Proteinen sowie aus Mineralien, Spurenelementen und Vitaminen essenziell. ...

Ein haariger Fall mit irreversiblen Folgen
Bestimmte Formen von Alopezie scheinen in jüngster Zeit explosionsartig zuzunehmen, wobei die genauen Ursachen bislang noch nicht vollständig geklärt sind. Handelt es sich dabei um eine ...

Die menschliche Haut in der modernen Kunst
Dr. Ralph Ubl, Professor für neuere Kunstgeschichte an der Universität Basel, stellte sich der schwierigen Herausforderung, einem Raum voller erwartungsvoller Dermatologen das Organ Haut ...