
Pneumopathie d’hypersensibilité
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Une nouvelle terminologie, un questionnaire spécifique visant à clarifier les cas suspects en Suisse et des options de traitement pharmacologique limitées, telles ont été les nouveautés concernant la pneumopathie d’hypersensibilité lors du congrès de la SSP de cette année à Lucerne.
La pneumopathie d’hypersensibilité (PH) représente environ 10% de toutes les pneumopathies interstitielles (PI). En Suisse, env. 200 personnes sont concernées par une PH chronique fibrosante ou non fibrosante. Le nombre de personnes souffrant d’une PH aiguë est nettement plus élevé. Alors que la PH aiguë est déclenchée chez les personnes préalablement sensibilisées par une exposition (généralement) de plusieurs heures à des quantités modérées à élevées d’allergènes, la PH chronique se développe à la suite d’une exposition à de faibles niveaux d’allergènes pendant des semaines, voire des années.1 Les résultats d’études plus anciennes indiquaient un pronostic plus favorable de la PH chronique par rapport aux autres PI.2 Des données plus récentes montrent désormais que l’espérance de vie dépend principalement de la présence d’une fibrose parenchymateuse. Ainsi, la durée médiane de survie des patients atteints de PH non fibrosante était d’environ 15 ans, contre 8 ans pour les patients atteints de PH fibrosante. Dans le cas d’une PH fibrosante avec «honeycombing», la durée médiane de survie est d’environ 2 à 3 ans et donc comparable à celle d’une fibrose pulmonaire idiopathique (FPI).3 Les nouvelles données d’études ont influencé la terminologie utilisée dans les directives actuelles sur la PH: au lieu de la stratification actuelle en PH aiguë, subaiguë et chronique, la PH est désormais divisée en PH fibrosante et non fibrosante.
Diagnostic multidisciplinaire préférable
Le diagnostic, en particulier de la PH fibrosante, s’avère difficile en raison de la diversité des symptômes et des nombreux chevauchements avec d’autres PI. Il est important d’identifier les facteurs déclencheurs liés à la PH à l’aide de l’anamnèse et d’entretiens structurés avec les patients. Une enquête Delphi auprès d’experts a permis d’établir une liste de déclencheurs importants (Fig.1).4 Pour clarifier les cas suspects en Suisse, le «Special Interest Group Interstital And Rare Lung Diseases» de la Société Suisse de Pneumologie a conçu un questionnaire avec des déclencheurs pertinents, disponible en plusieurs langues. De surcroît, la HRCT («high-resolution computed tomography») et la bronchoscopie, y compris la biopsie pulmonaire et le lavage broncho-alvéolaire (LBA), fournissent des indications importantes. La détermination d’anticorps IgG spécifiques (précipitines) est également souvent discutée. Les lignes directrices ne recommandent pas une telle procédure en raison de sa faible spécificité. «La détection des précipitines peut faciliter le diagnostic, mais elle ne suffit pas à elle seule pour diagnostiquer une PH», a déclaré la PD Dre méd. Sabina Guler, de l’Hôpital universitaire, Hôpital de l’Île, à Berne. La réalisation d’un test de provocation bronchique dans le cadre d’un diagnostic de routine n’est pas non plus recommandée.
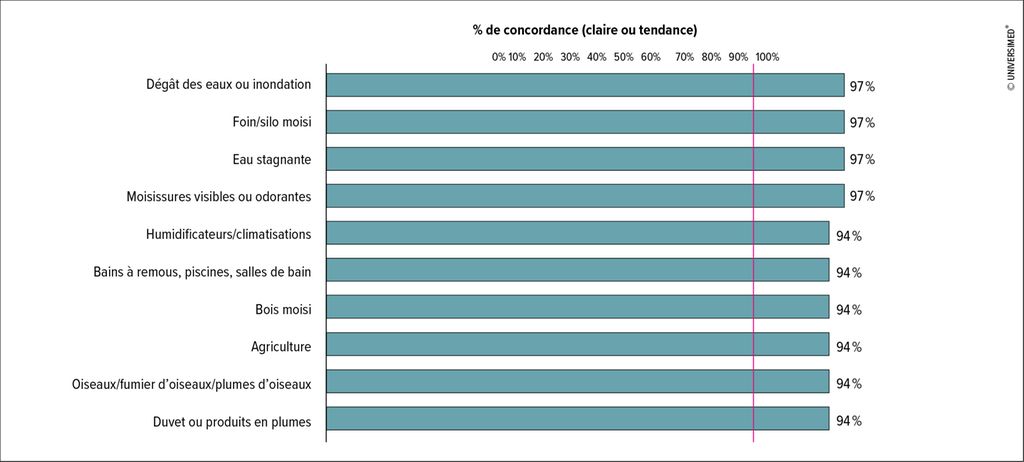
Fig. 1: Quelles sont les expositions pertinentes? (adapté d’après Barnes et al.)4
«L’idéal est de poser le diagnostic dans le cadre d’une discussion multidisciplinaire intégrant les différentes pièces du puzzle», a déclaré la spécialiste. Si aucune équipe multidisciplinaire n’est disponible, un algorithme avec des critères diagnostiques peut être utile pour le diagnostic.5 L’algorithme présente une bonne performance avec une certitude diagnostique de ≥70% (sensibilité: 0,74; spécificité: 0,9). Par rapport à l’équipe multidisciplinaire, l’algorithme était toutefois inférieur en termes de certitude diagnostique, comme l’a montré une étude de l’intervenante.6 «Le diagnostic multidisciplinaire est actuellement ce que nous avons de mieux», a déclaré S.Guler.
Absence de lignes directrices de traitement
Jusqu’à présent, il n’existe pas de recommandations officielles de traitement pour la PH. De plus, il n’existe que peu d’études cliniques évaluant un traitement pharmacologique en cas de PH. «Une mesure importante consiste à éliminer le déclencheur», a déclaré le PD Dr méd. Christian Clarenbach, de l’Hôpital universitaire de Zurich. Chez les patients atteints de PH active, cette approche entraîne une récupération visible du tissu pulmonaire ainsi qu’une amélioration de la fonction respiratoire et soutient le diagnostic. Les mesures d’éviction ont un effet nettement plus faible, voire nul, chez les patients atteints de PH chronique fibrosante.
Traitements immunosuppresseurs
Les études sur les immunosuppresseurs donnent des résultats controversés. En ce qui concerne le traitement par corticostéroïdes, une seule petite étude randomisée a été menée en 1992 auprès de 36 patients atteints de PH active.7 Ils ont été traités pendant 8 semaines soit par 5mg de prednisolone par jour, soit par un placebo. Un mois après le début du traitement, les résultats ont montré que la fonction pulmonaire se rétablissait plus rapidement sous corticothérapie que sous placebo.
Deux études rétrospectives ont évalué l’effet d’un traitement immunosuppresseur par mycophénolate mofétil (MMF) ou azathioprine (AZA). La première étude a porté sur des patients atteints de PH chronique et recevant un traitement de fond à base de prednisone (12mg/j). Après une durée médiane de traitement d’un an, aucune augmentation nette de la capacité vitale (CVF) n’a été observée suite à l’ajout du MMF ou de l’AZA. En revanche, la capacité de diffusion (DLCO) s’est améliorée de manière significative de 4,2% sous traitement (p<0,001).8 La deuxième étude a porté sur une majorité de patients souffrant de PH chronique et traités soit par prednisone, soit par MFF, soit par AZA. Aucune différence n’a été observée entre les traitements comparés en termes de CVF.9 Il est intéressant de noter que les patients de cette étude qui n’ont pas été traités par immunosuppresseurs ont vécu plus longtemps que ceux qui ont reçu un traitement immunosuppresseur.
Le rituximab, un anticorps anti-CD20 utilisé avec succès dans le traitement de la PI associée à la collagénose, pourrait également constituer une option de traitement. Les deux études disponibles à ce jour sur le rituximab dans la PH sont toutefois trop peu concluantes pour pouvoir en tirer une recommandation de traitement, en raison du faible nombre de patients (n<10).
Traitement antifibrotique
Ces dernières années, plusieurs études ont été publiées pour évaluer l’impact d’un traitement antifibrotique par nintédanib chez les patients atteints de PI. Après que les études INPULSIS-1 et -2 avaient mis en évidence une diminution de la CVF d’environ la moitié par rapport au placebo chez les patients atteints de FPI, un effet comparable a également été démontré dans les études INBUILD-1 et -2 chez les patients atteints de PI fibrosante et dans l’étude SENSCIS pour la PI associée à la sclérodermie.11 Une analyse de sous-groupe des études INBUILD a montré que le traitement par nintédanib entraînait également une diminution retardée de la CVF chez les patients atteints de HP fibrosante chronique.12
Transplantation du poumon
«Pour les jeunes patients atteints de PH sans comorbidités pertinentes, qui ne répondent pas au traitement pharmacologique et dont la fonction pulmonaire continue de décliner, la transplantation pulmonaire pourrait être une option thérapeutique», a déclaré C. Clarenbach. Les indications les plus fréquentes pour une transplantation pulmonaire sont la FPI (30%), suivie de la BPCO (26%) et de la fibrose kystique (13%). Selon lui, il est important d’orienter les patients atteints de PI à un stade précoce.
Source:
Congrès annuel de la Société Suisse de Pneumologie (SSP), 30 mars au 1er avril 2022, Lucerne
Littérature:
1 Costabel U et al.: Hypersensitivity pneumonitis. Nat Rev Dis Primers 2020; 6: 65 2 Ryerson CJ et al.: Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest 2014; 145: 723-8 3 Salisbury ML et al.: Hypersensitivity pneumonitis: radiologic phenotypes are associated with distinct survival time and pulmonary function trajectory. Chest 2019; 155: 699-711 4 Barnes H et al.: A systematically derived exposure assessment instrument for chronic hypersensitivity pneumonitis. Chest 2020; 157: 1506-12 5 Morisset J et al.: Identification of diagnostic criteria for chronic hypersensitivity pneumonitis: an international modified delphi survey. Am J Resp Crit Care Med 2018; 197: 1036-44 6 Guler SA et al.: Performance of a diagnostic algorithm for fibrotic hypersensitivity pneumonitis. A case-control study. Respir Res 2021; 22: 120 7 Kokkarinen JI et al.: Effect of corticosteroid treatment on the recovery of pulmonary function in farmer‘s lung. Am Rev Respir Dis 1992; 145: 3-5 8 Morisset J et al.: Use of mycophenolate mofetil or azathioprine for the management of chronic hypersensitivity pneumonitis. Chest 2017; 151: 619-25 9 Adegunsoye A et al.: Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res 2017; 3: 00016-2017 10 Morell F et al.: Addition of rituximab to oral corticosteroids in the treatment of chronic hypersensitivity pneumonitis. Arch Bronconeumol 2020; 56: 255-6 11 Bonella F et al.: Meta-Analysis of effect of nintedanib on reducing FVC decline across interstitial lung diseases. Adv Ther 2022; 39: 3392-402 12 Wells AU et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med 2020; 8: 453-60
Das könnte Sie auch interessieren:

BPCO: identification de patients non diagnostiqués
De nombreuses personnes atteintes de BPCO et/ou d’asthme n’ont jamais été diagnostiquées et ne sont donc pas traitées. Plusieurs études publiées ces derniers mois se penchent sur ce ...

Diarrhée chronique: déroulement de l’examen
La diarrhée chronique est le symptôme de différentes maladies. Le Pr Alain Schoepfer a expliqué comment rechercher au mieux le facteur déclenchant dans son exposé lors du congrès annuel ...

Réduction, voire arrêt des corticoïdes grâce à la biothérapie?
Les corticoïdes oraux constituent le traitement de référence des exacerbations de l’asthme et sont également utilisés pour celles de la BPCO. Cette norme s’appuie toutefois sur des ...