
Le rôle central des bactéries peu abondantes dans le microbiome des voies aériennes dans la petite enfance
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Le microbiome pulmonaire joue un rôle majeur dans la santé des voies aériennes, notamment dans des maladies telles que la mucoviscidose. L’outil logiciel raspir, récemment développé, permet de déterminer quasiment toutes les espèces d’une communauté bactérienne – les résultats d’une analyse in silico de données du métagénome de patients pédiatriques atteints de mucoviscidose illustrent leur importance. Grâce à cet outil, des facteurs importants qui influencent l’ensemble du microbiome, comme par exemple une approche de traitement probiotique, pourront être étudiés en détail à l’avenir.
Keypoints
-
Jusqu’à présent, il n’a pas été possible de déchiffrer de manière taxonomique les espèces bactériennes présentes en petit nombre dans les analyses du microbiome basées sur le séquençage.
-
L’outil logicielraspirréduit le signal interférent associé à la méthode et permet ainsi de déterminer quasiment toutes les espèces d’une communauté bactérienne.
-
Les bactéries présentes en petites quantités dans les voies aériennes stabilisent le réseau du microbiome pulmonaire sain.
-
Les approches de traitement probiotiques avec une grande diversité bactérienne et une faible dominance d’espèces bactériennes individuelles pourraient stabiliser le réseau bactérien sous-développé des voies aériennes dans le contexte des maladies pulmonaires chroniques.
Dans le cadre d’un diagnostic microbien indépendant de la culture, l’ADN complet d’un échantillon de patient est isolé, fragmenté, séquencé et les fragments d’ADN sont mis en correspondance avec une base de données de génomes bactériens de référence. L’objectif est de déterminer la communauté microbienne dans un organe tel que les voies aériennes. Le problème est que de nombreux microbes partagent de courtes séquences génétiques avec des microbes de leur communauté et avec d’autres entrées de la base de données de référence. Une mise en correspondance incorrecte des fragments d’ADN biologique avec les séquences homologues de la base de données de référence entraîne une détection fausse positive d’espèces bactériennes qui n’existent pas réellement dans l’échantillon. Afin de minimiser la proportion de ces résultats faux positifs, les pipelines d’évaluation ont fixé une valeur seuil afin d’émettre seulement des affirmations sur les bactéries très abondantes présentes avec certitude, c’est-à-dire les bactéries qui représentent de façon cumulée une proportion de 90–99% de la communauté. Cependant, des informations importantes sur les espèces peu abondantes sont ainsi perdues.
Le microbiome humain est composé d’un petit nombre d’espèces bactériennes très abondantes et d’un grand nombre d’espèces peu abondantes.1 On peut donc supposer que les bactéries peu abondantes déterminent la majorité de la diversité génétique et de la flexibilité fonctionnelle du microbiome.2 L’élimination de ces espèces bactériennes au début de l’analyse des données se traduit donc par une image incomplète de la communauté microbienne du corps humain.
Développement d’un outil logiciel pour déterminer les bactéries très abondantes et peu abondantes à partir d’échantillons complexes de métagénome
Nous avons récemment publié un outil logiciel («rare species identifier»; raspir) qui examine la distribution des fragments le long du génome, ou l’organisation du génome des espèces bactériennes.3 Si l’on compare les bactéries de la même espèce, la disposition des gènes dans le génome est similaire. Étant donné que l’ADN est reproduit et séquencé de manière aléatoire dans le séquençage profond, les fragments d’ADN de toutes les parties du génome bactérien devraient être détectables dans les espèces bactériennes effectivement présentes. En revanche, les segments de séquence faux positifs ne sont pas répartis uniformément sur l’ensemble du génome. En conséquence, une astuce mathématique peut être utilisée pour distinguer les espèces bactériennes très abondantes et peu abondantes des résultats faux positifs, selon les modalités suivantes: on calcule la distance entre tous les fragments d’ADN attribués à un génome de référence, on décompose le signal de position en ses composantes spectrales et on compare le signal dans la gamme de fréquence avec un signal de référence théorique simulant une distribution égale idéale des fragments d’ADN.3 La Figure 1 explique le principe.
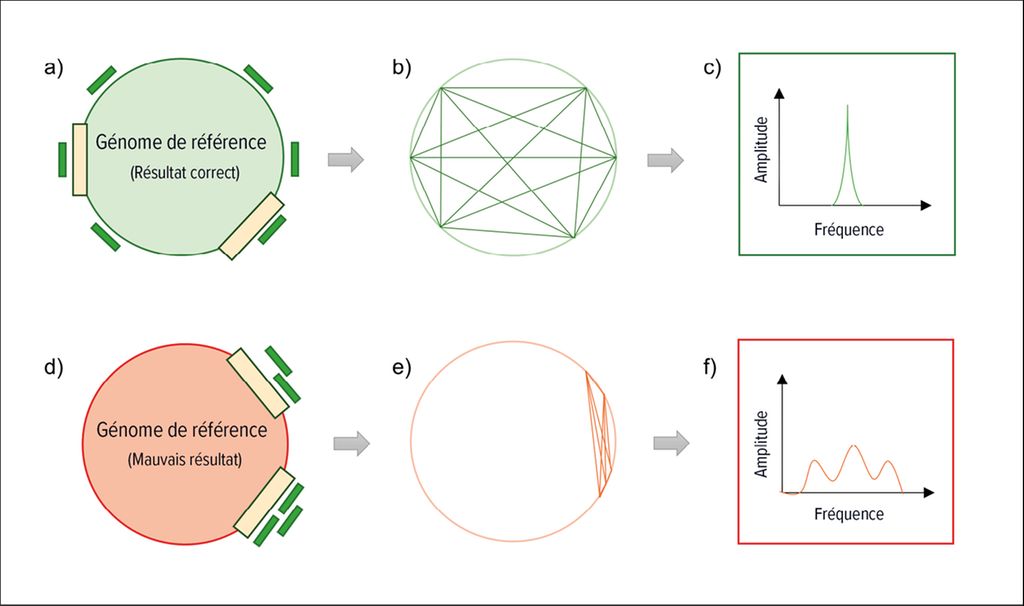
Fig. 1: Illustration simplifiée de la manière d’utiliser le placement de morceaux d’ADN le long d’un génome de référence pour détecter la présence d’une bactérie dans l’échantillon du patient. (a)Lorsque les fragments d’ADN biologique (en vert) sont attribués au génome de la bactérie présente, les morceaux d’ADN se répartissent uniformément sur le génome. (b) La distance entre tous les fragments assignés au génome de référence correct est calculée. (c) Le signal de position est décomposé en ses composantes spectrales et comparé à un signal de référence similaire au signal de position présenté ici. (d) Si les fragments d’ADN (verts) sont assignés au mauvais génome (rouge), c’est-à-dire à une bactérie qui n’était pas dans l’échantillon mais qui partage des séquences génétiques avec la bactérie correcte (barres jaunes), les morceaux de fragments se regroupent au niveau de ces sections. (e) La distance entre tous les fragments est calculée. (f) Le signal est décomposé en ses composantes spectrales et comparé à un signal de référence similaire au signal de la figure c
Résultats d’une analyse in silico de données métagéniques précédemment publiées
Pour analyser l’importance des bactéries à faible abondance dans le développement précoce du métagénome des voies aériennes chez les enfants en bonne santé et ceux atteints de mucoviscidose entre 0 et 6 ans, nous avons utilisé les données primaires publiées précédemment dans le cadre d’une étude clinique.4 Une nouvelle analyse avec raspir3 a révélé que chez les enfants en bonne santé, la plupart des bactéries à abondance élevée et faible formaient un réseau de fond persistant (Fig.2, à gauche). Les bactéries de fond étaient donc déjà détectables au cours de la première année de la vie et restaient présentes chez les tout-petits et à l’âge préscolaire. En fonction du stade de développement de l’enfant, d’autres bactéries non persistantes sont apparues dans les voies aériennes. Chez les jeunes enfants atteints de mucoviscidose, en revanche, seules quelques bactéries persistantes abondantes et peu abondantes ont été détectées dans le fond (Fig. 2, à droite). La plupart des bactéries peu abondantes étaient affectables à la petite enfance (2–3 ans). On peut en conclure que le système immunitaire des enfants en bonne santé est continuellement stimulé par un réseau persistant distinct de bactéries commensales des voies aériennes, tandis que le système immunitaire des jeunes enfants atteints de mucoviscidose est largement entraîné par les bactéries non persistantes et, plus tard, par les pathogènes de la mucoviscidose.5 Pour identifier les variables qui distinguent un métagénome des voies aériennes sain d’un métagénome de mucoviscidose, nous avons utilisé un algorithme de classification par forêts d’arbres décisionnels basé sur les composants du métagénome des voies aériennes (bactéries abondantes et peu abondantes) et des variables spécifiques à l’hôte (âge, indice de masse corporelle, indice de clairance pulmonaire, insuffisance pancréatique contre suffisance pancréatique). Il a été constaté que plus de 70% des variables utilisées pour le processus de décision («sain» ou «mucoviscidose») étaient associées aux bactéries peu abondantes).
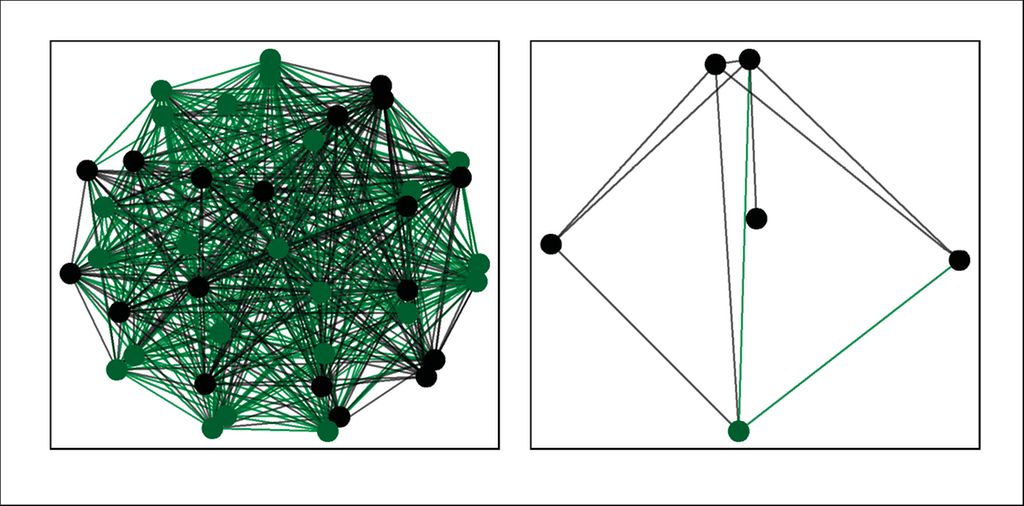
Fig. 2: Le réseau de fond persistant formé par les bactéries peu abondantes (en vert) et abondantes (en noir) chez des enfants sains (à gauche) et atteints de mucoviscidose (à droite)
Si les bactéries abondantes ont joué un rôle mineur dans le processus de décision de classification, l’influence des facteurs spécifiques à l’hôte était négligeable. Àl’aide de l’apprentissage automatique, nous avons simulé des approches de traitement probiotiques en introduisant un nombre croissant de bactéries très abondantes et peu abondantes issues du réseau sain dans le réseau de fond sous-développé de la mucoviscidose et comparé les structures graphiques nouvellement formées avec celles du réseau sain. Il s’est avéré que le réseau bactérien de la mucoviscidose a gagné en stabilité et en résilience lorsque des bactéries saines présentant une grande diversité d’espèces et une faible dominance d’espèces individuelles ont été transférées dans le réseau. Cependant, le réseau de la mucoviscidose d’origine a perdu de sa stabilité lorsque des bactéries commensales individuelles à fréquence élevée ont été incorporées dans le réseau de la mucoviscidose.
Perspective
Les espèces bactériennes présentes en petite quantité jouent un rôle central dans le microbiome des voies aériennes saines et malades et devraient donc être prises en compte dans les futures analyses du microbiome. Nos études de simulation in silico basées sur la corrélation fournissent une première indication que, selon le type d’approche de traitement probiotique, le réseau de base déjà sous-développé des bactéries commensales de la mucoviscidose pourrait être stabilisé mais aussi déstabilisé davantage. L’étape suivante consiste à utiliser des modèles complexes in vitro et in vivo pour tester la causalité de nos analyses.
Littérature:
1 Ma ZS: Power law analysis of the human microbiome. Mol Ecol 2015; 24: 5428-45 2 Jousset A et al.: Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J 2017; 11: 853-62 3 Pust MM, Tümmler B: Identification of core and rare species in metagenome samples based on shotgun metagenomic sequencing, Fourier transforms and spectral comparisons. ISME COMMUN 2021; 1: 2 4 Pust MM et al.: The human respiratory tract microbial community structures in healthy and cystic fibrosis infants. NPJ Biofilms Microbiomes 2020; 6: 61 5 Coburn B et al.: Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep 2015; 5: 10241
Das könnte Sie auch interessieren:

Modulateurs du récepteur de la sphingosine-1-phosphate dans le traitement de la SEP
Les modulateurs du récepteur de la sphingosine-1-phosphate représentent une classe de médicaments prometteurs pour le traitement de deuxième ligne de la sclérose en plaques (SEP). Deux ...

Présentation des données pertinentes pour la pratique sur le mavacamten dans la CMHO
Le mavacamten, un inhibiteur de la myosine, a été la première substance de cette classe à être autorisée en 2023 pour le traitement de la cardiomyopathie hypertrophique obstructive (CMHO ...

Hypertension secondaire: formes, diagnostic & options thérapeutiques
La fréquence d’une cause secondaire d’hypertension artérielle est de 10%. En cas de suspicion d’hypertension secondaire, il convient de procéder à un examen ciblé afin de pouvoir, dans l ...