
De plus en plus d’options thérapeutiques dans l’amylose cardiaque
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Alors que l’évolution de l’amylose cardiaque était d’issue fatale, progressive et non influençable par un traitement il y a une dizaine d’années, il existe aujourd’hui plusieurs traitements spécifiques à l’amylose. Certains sont déjà autorisés ou ont passé avec succès les études de phase III. Le coût élevé de ces traitements constituera à l’avenir un enjeu majeur dans la pratique clinique quotidienne.
Keypoints
-
L’amylose ATTR avec cardiomyopathie se caractérise par le dépôt de variants anormaux de transthyrétine dans le cœur.
-
Il n’existe aucune preuve issue d’études prospectives démontrant l’efficacité des traitements conventionnels contre l’insuffisance cardiaque en cas d’amylose ATTR avec cardiopathie.
-
Depuis l’autorisation du tafamidis dans l’indication amylose ATTR avec cardiomyopathie, d’autres traitements spécifiques à l’amylose ont été utilisés avec succès dans des études.
-
Dans ses dernières directives sur les cardiomyopathies, l’ESC fournit des recommandations détaillées sur le diagnostic de l’insuffisance cardiaque due à l’amylose cardiaque.
L’amylose cardiaque est une cardiomyopathie infiltrante évolutive qui se caractérise par le dépôt de protéines mal repliées dans l’interstitium du myocarde associé à une hypertrophie myocardique afférente. Il s’ensuit une altération de la fonction ventriculaire gauche diastolique et, ultérieurement, de la fonction ventriculaire gauche systolique, qui se manifeste cliniquement par une insuffisance cardiaque associée à une dyspnée d’effort, à des œdèmes des jambes et à un épuisement rapide. Les principales formes sont l’amylose à chaînes légères (AL), souvent associée aux lymphomes non hodgkiniens, et l’amylose à transthyrétine (ATTR). Cette dernière se distingue par le dépôt de variants anormaux de la protéine de transport transthyrétine dans le cœur.
«Les options thérapeutiques dans l’amylose cardiaque ont considérablement évolué au cours des dix dernières années», a expliqué le PD Dr méd. Franz Duca, de l’Université de médecine de Vienne, lors du Wiener Kongress Kardiologie, en soulignant que des traitements spécifiques à l’amylose sont disponibles depuis quelques années et qu’ils permettent d’intervenir de manière causale dans l’évolution de la maladie. Par ailleurs, les traitements conventionnels contre l’insuffisance cardiaque ne doivent pas être totalement négligés en cas d’amylose cardiaque.
Preuves très limitées pour les traitements conventionnels
En ce qui concerne les traitements conventionnels, une analyse rétrospective portant sur plus de 2000 patient·es atteint·es d’amylose cardiaque ATTR suggère que les antagonistes du récepteur des minéralocorticoïdes (ARM) sont associés à une réduction de la mortalité dans la population totale ainsi que dans un sous-groupe dont la fraction d’éjection ventriculaire gauche (FEVG) est <40%. Les bêtabloquants à faible dose ont uniquement réduit la mortalité chez les patient·es dont la FEVG était <40%. En revanche, aucune réduction de la mortalité n’a été démontrée pour l’inhibition du SRAA par des inhibiteurs de l’ECA (iECA) ou des antagonistes des récepteurs de l’angiotensine (ARA). Les iECA/ARA ont également été arrêtés plus fréquemment que les autres substances étudiées.1 Une étude de plus petite taille portant sur des patient·es atteint·es d’amylose ATTR avec cardiomyopathie a même montré une réduction du risque de mortalité de l’ordre de 70% sous bêtabloquants. Cependant, 25% des patient·es ont dû interrompre le traitement en raison d’une intolérance.2 Comme il s’agit exclusivement de données rétrospectives, ces travaux peuvent au mieux permettre de formuler des hypothèses, a expliqué F. Duca. D’une manière générale, on évolue dans une «zone sans preuve» pour les traitements de soutien. Les indications relatives aux bêtabloquants et aux ARM doivent donc être accompagnées de la mention «efficacité possible». De même, il y a des signes de réduction de la mortalité sous inhibiteurs du SGLT2 (iSGLT2), pour lesquels seules des données rétrospectives sont disponibles.3
Pour les diurétiques de l’anse, un lien entre des doses plus élevées et une augmentation de la mortalité a été démontré. «Cela s’explique par le simple fait que les patient·es gravement atteint·es ont besoin de plus de diurétiques», explique F. Duca. Dans la pratique clinique, un besoin croissant en diurétiques peut être considéré comme un simple indicateur d’aggravation de la maladie. Il ne faut pas oublier que l’amylose présente généralement un risque de thrombose nettement accru et indépendant du score CHA2DS2-VASc, qui reste élevé même sous anticoagulants. De même, des arythmies surviennent chez plus de la moitié des personnes concernées. Aucune donnée concernant une éventuelle influence sur la survie n’est toutefois disponible.
Traitements causaux déjà utilisés dans l’amylose ATTR
Les traitements spécifiques à l’amylose ont connu un développement rapide en un peu plus d’une décennie. Le tafamidis, un stabilisateur de la TTR qui inhibe la dissociation de la TTR en monomères, constitue une option thérapeutique dans l’amylose ATTR. L’étude de phase III ATTR-ACT portant sur le tafamidis a débuté en 2013 et a montré un avantage significatif en termes de survie par rapport au placebo, avec une amélioration de la qualité de vie. L’utilisation à un stade précoce de la maladie semble avantageuse, car les patient·es du groupe sous placebo ont certes tiré profit d’un passage au tafamidis, mais le désavantage déjà existant n’a pas pu être rattrapé. En outre, F. Duca a souligné que les courbes du placebo et du traitement actif ne s’étaient séparées qu’après environ 18 mois dans l’étude ATTR-ACT (Fig.1). Il faut donc informer les patient·es qu’il ne faut pas s’attendre à un effet rapide. L’étude ATTR-ACT a conduit à l’autorisation du tafamidis en 2019.4
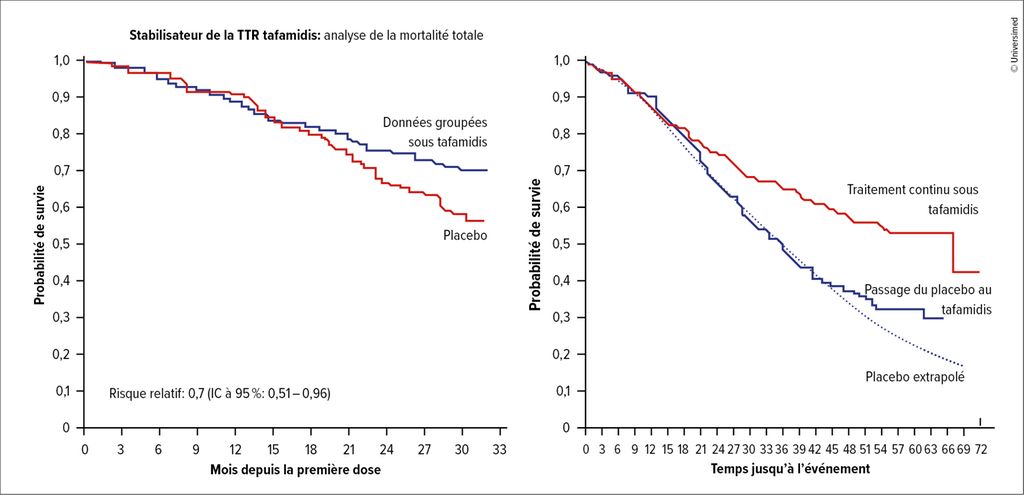
Fig.1: Probabilité de survie sous tafamidis (modifié selon Maurer MS et al. 2018)4
L’acoramidis est un autre stabilisateur de la TTR pour lequel une stabilisation de la TTR de plus de 90% a été démontrée exvivo. Dans l’étude d’autorisation ATTRibute-CM, l’acoramidis a réduit de manière significative et nette plusieurs critères d’évaluation combinés, dont la mortalité totale, les hospitalisations pour causes cardiovasculaires, la diminution de la distance parcourue au test de marche de 6 minutes et la variation du taux de NT-proBNP.5
Une autre stratégie thérapeutique dans l’amylose ATTR est l’inhibition pharmacologique de la synthèse de la TTR dans le foie. La molécule antisens inotersen et le «small interfering RNA» (siRNA) patisiran ont été développés à cet effet. Ces deux molécules, tout comme le siRNA vutrisiran, sont autorisées dans l’indication amylose ATTR avec neuropathie.
Dans l’indication amylose AL, le daratumumab, un anticorps monoclonal humain anti-CD38, une glycoprotéine exprimée notamment à la surface des plasmocytes, a été examiné dans l’étude ANDROMEDA. Il a entraîné une meilleure réponse hématologique ainsi qu’une réduction des lésions organiques par rapport au placebo.6
Bonnes données également disponibles pour le siRNA vutrisiran dans l’amylose cardiaque ATTR
Le vutrisiran a lui aussi déjà été étudié dans l’indication amylose ATTR avec cardiomyopathie. L’étude HELIOS-B a révélé une significativité statistique pour les critères d’évaluation primaires et secondaires dans la population totale et dans la population sous monothérapie, c’est-à-dire chez les patient·es qui n’étaient pas traité·es par le tafamidis à l’inclusion. Les réductions de la mortalité totale et des événements cardiovasculaires récurrents ont été de 28% dans la population totale et de 33% dans la population sous monothérapie. La diminution de la distance parcourue au test de marche de 6 minutes et du score KCCQ-OS (Kansas City Cardiomyopathy Questionnaire Overall Summary) a également été freinée de manière significative sous vutrisiran. F. Duca a souligné que 40% de la population de l’étude HELIOS B étaient déjà sous tafamidis, et a notamment mis l’accent sur la réduction des hospitalisations, qui est subjectivement perçue comme plus importante que la seule augmentation de l’espérance de vie par de nombreuses personnes concernées, pour la plupart âgées.7
Au fil des années, les études mettent en évidence une amélioration générale du pronostic de la maladie. Le groupe sous placebo de l’étude HELIOS B a obtenu de meilleurs résultats que le groupe sous traitement actif de l’étude ATTR-ACT, ce qui s’explique principalement par le fait que les patient·es atteint·es d’amylose ATTR avec cardiomyopathie sont aujourd’hui diagnostiqué·es et traité·es plus tôt. «Ce ne sont plus les patient·es que nous voyions il y a dix ans», déclare F. Duca. En conséquence, il est difficile de comparer l’efficacité des premiers traitements autorisés dans l’amylose ATTR avec celle des nouvelles substances.
Perspectives possibles:
ciseaux génétiques, «depleters» et coûts thérapeutiques élevés
Un espoir pour l’avenir dans ce domaine est un traitement basé sur les ciseaux génétiques CRISPR-Cas9, qui pourrait mettre complètement fin aux dépôts amyloïdes. Des études sont actuellement menées chez l’humain. Une seule application du traitement par «gene-editing» NTLA-2001 a permis de réduire les taux sériques de la TTR d’environ 90%.8 Une autre approche innovante consiste à améliorer la dégradation des dépôts amyloïdes. Plusieurs «depleters» aux mécanismes d’action différents font déjà l’objet d’études chez l’humain. En cas d’autorisations, cela offrirait de nouvelles options de traitements combinés ou séquentiels.
«La question des coûts constituera à l’avenir un enjeu majeur. Le tafamidis était déjà, et de loin, le médicament le plus cher disponible», a déclaré F. Duca. Les nouvelles substances déjà utilisées en neurologie sont encore nettement plus chères. Ces prix peuvent être pratiqués, car les amyloses sont considérées comme des maladies rares par les organismes de réglementation. Il n’est pas certain que cette estimation se confirmera à l’avenir, et il existe un risque futur que les personnes concernées se voient refuser ce traitement efficace en raison des coûts.
Dans ce contexte, le PD Dr méd. Andreas Kammerlander de l’Université de médecine de Vienne a souligné l’absence d’une définition généralement acceptée des maladies rares. La plupart du temps, on qualifie de rares les maladies dont la prévalence est ≤1:2000. Les cardiopathies rares ne sont donc pas aussi rares qu’on pourrait le croire. Selon Orphanet, le choc cardiogénique entre par exemple dans cette catégorie. Depuis 2023, les maladies rares sont également prises en compte pour la première fois de manière explicite dans les directives de l’ESC. Elles traitent notamment en détail les amyloses cardiaques et indiquent un parcours diagnostique recommandé, qui prévoit également des méthodes diagnostiques non invasives pour l’amylose ATTR (Fig.2). Les directives soulignent aussi l’absence de preuves pour l’utilisation de médicaments conventionnels contre l’insuffisance cardiaque en cas d’amylose cardiaque. Pour l’amylose ATTR, il est recommandé de stabiliser la transthyrétine ainsi que de réduire sa production. Cela signifie qu’il est conseillé d’utiliser le tafamidis le plus tôt possible dans l’évolution de la maladie (pour les classes I et II de la NYHA). Le patisiran et l’inotersen sont par ailleurs déjà mentionnés, avec une mention précisant qu’ils sont actuellement uniquement autorisés dans l’amylose ATTR avec polyneuropathie et avec ou sans cardiomyopathie.
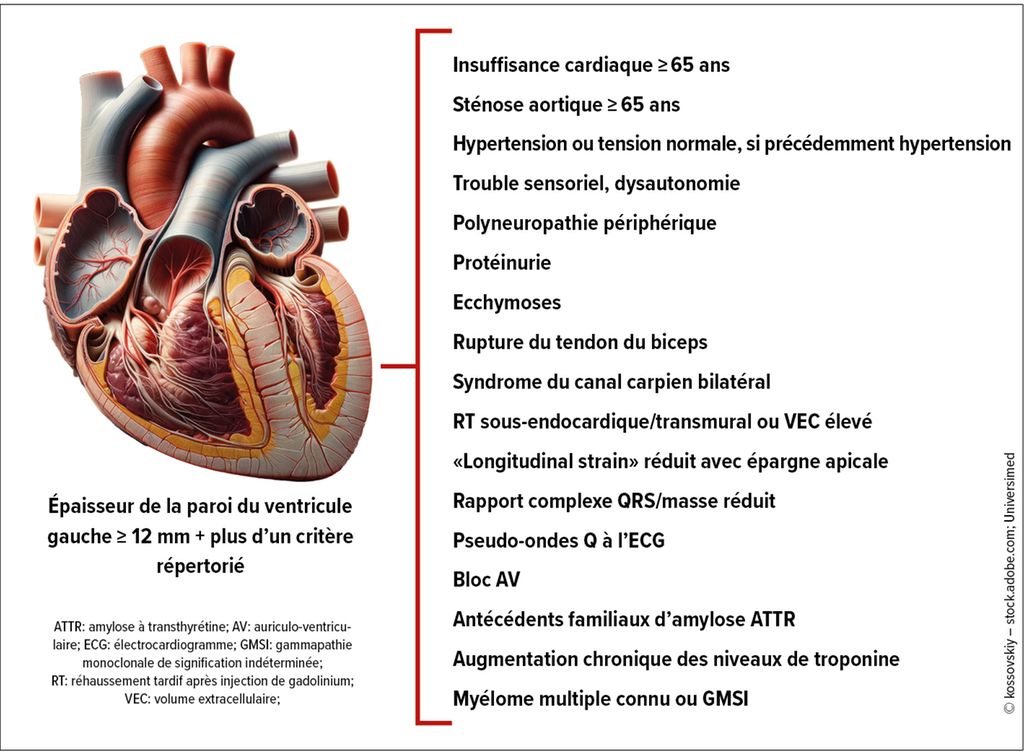
Fig.2: Dépistage de l’amylose cardiaque
Source:
Présentation «Rare diseases» lors de la 7e session principale du Wiener Kongress Kardiologie s’étant tenu le 19 octobre 2024 à Vienne
Littérature:
1 Ioannou A et al.: Conventional heart failure therapy in cardiac ATTR amyloidosis. Eur Heart J 2023; 14; 44: 2893-907 2 Barge-Caballero G et al.: Beta-blocker exposure and survival in patients with Transthyretin amyloid cardiomyopathy. Mayo Clin Proc 2022; 97: 261-73 3 Porcari A et al.: SGLT2 inhibitor therapy in patients with Transthyretin amyloid cardiomyopathy. J Am Coll Cardiol 2024; 83: 2411-22 4 Maurer MS et al.: Tafamidis treatment for patients with Transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007-16 5 Gillmore JD et al.: Efficacy and safety of Acoramidis in Transthyretin amyloid cardiomyopathy. N Engl J Med 2024; 390: 132-42 6 Kastritis E et al.: Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med 2021; 385: 46-58 7 Fontana M et al.: Vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. N Engl J Med 2025; 392: 33-44 8 Gillmore JD et al.: CRISPR-Cas9 in vivo gene editing for Transthyretin amyloidosis. N Engl J Med 2021; 385: 493-502 9 Arbelo E et al.: 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J 2023; 44: 3503-626
Das könnte Sie auch interessieren:

Une voix forte pour la santé mondiale
Lors du congrès de printemps de la Société Suisse de Médecine Interne Générale (SSMIG), Ilona Kickbusch, sociologue et politologue de renom, s’est exprimée sur la santé mondiale. Il est ...

«Planetary Health»: l’interaction entre la santé et le changement climatique
Le changement climatique est une réalité depuis longtemps. En Suisse, les dix dernières années ont été 2,5°C plus chaudes que la moyenne pendant la période préindustrielle et les ...
Traitement hypolipémiant chez les personnes vivant avec le VIH
Les personnes vivant avec le virus de l’immunodéficience humaine (VIH) présentent un risque accru de maladies cardiovasculaires athéroscléreuses. De plus, le traitement antirétroviral et ...