
CAR-T-Zell-Therapie bei soliden Tumoren
Während Anti-CD19-chimäre-Antigenrezeptor(CAR)-T-Zellen in den letzten Jahren Einzug in das hämatoonkologische Behandlungsspektrum erhalten haben, gibt es bislang keine ausreichende Evidenz, dass auch Patienten, die an soliden Tumoren leiden, von CAR-T-Zellen profitieren können. Auf der 7th Immunotherapy of Cancer Conference (ITOC) in München werden in einem translationalen Umfeld aktuelle Schwierigkeiten und Lösungsansätze zum Thema „adoptive T-Zell-Therapie für solide Tumoren“ diskutiert.
Die CAR-T-Zell-Therapie beruht auf einem Ex-vivo-Gentransfer eines synthetischen Antigenrezeptors in T-Zellen des Patienten. Hohe Ansprechraten gegen refraktäre akute lymphatische Leukämie und hochmaligne B-Zell-Lymphome führten 2017 (FDA) beziehungsweise 2018 (EMA) zur ersten Zulassung der neuen Behandlungsart in Form von Anti-CD19-CAR-T-Zellen.1–5 Aktuelle Bemühungen zielen darauf ab, die Therapieerfolge der Anti-CD19-CAR-T-Zellen auf andere Tumorentitäten zu übertragen. Insbesondere gegen solide Tumoren ist die therapeutische Wirksamkeit des adoptiven T-Zell-Transfers nach wie vor begrenzt.6, 7
Resistenzmechanismen solider Tumoren gegen CAR-T-Zellen
Erschwerte Zellinfiltration
Eine hohe Anzahl an tumorinfiltrierenden Lymphozyten (TILs) korreliert in zahlreichen Tumorentitäten mit einer besseren Prognose.8–11 Um aus dem Blutstrom ins Gewebe einzutreten, kommunizieren Lymphozyten über Chemokine, Selectine und Integrine mit dem umliegenden Gewebe und dem Endothel.12 In soliden Tumoren wird die Infiltration endogener und transferierter zytotoxischer T-Zellen jedoch häufig durch eine anormale Vaskularisierung, eine dichte extrazelluläre Matrix und die Modulierung der Integrinexpression erschwert.13, 14
Auch die Expression von Chemokinen kann bedeutsam variieren, sodass es zur Rekrutierung von Immunzellen, die zur Tumorprogression beitragen, kommt. Beispielhaft zu nennen sind Ovarialkarzinome und Mammakarzinome, welche in der Lage sind, „CC-chemokine ligand 22“ (CCL22) und CCL28 überzuexprimieren, um die Migration regulatorischer T-Zellen (Tregs) in den Tumor zu verstärken.15, 16
Diese Gegebenheiten gezielt zu umgehen oder sogar auszunutzen stellt eine attraktive Möglichkeit dar, um die Infiltration adoptiv transferierter T-Zellen in den Tumor zu ermöglichen. Es gilt dementsprechend die zugrundeliegende Tumorbiologie zu verstehen und therapeutische Strategien zu entwickeln.
Limitierte Migration der CAR-T-Zellen in den Tumor kann durch intratumorale oder intrakompartimentale Applikation des
T-Zell-Produkts überwunden werden.17 Diese Strategie ist im klinischen Alltag jedoch häufig nicht anwendbar. Eleganter sind Ansätze,bei denen CAR-T-Zellen gentechnisch modifiziert werden, sodass Matrix-degradierende Enzyme oder Chemokinrezeptoren überexprimiert werden. Zahlreiche Studien in präklinischen Modellen demonstrieren die Umsetzbarkeit und Funktionalität derartiger Strategien.18, 19 Derzeit untersuchen zwei klinische Studien die therapeutische Wirksamkeit adoptiv transferierter T-Zellen mit Überexpression von Chemokinrezeptoren. In NCT01740557 wird die Wirksamkeit von CXC-Motiv-Chemokinrezeptor-2(CXCR2)-transduzierten autologen TILs als Therapie bei Patienten mit Melanom untersucht. In NCT03602157 werden Anti-CD30-CAR-T-Zellen zusätzlich mit C-C-Chemokinrezeptor Typ 4 (CCR4) transduziert, um eine Therapie von Patienten mit CD30+ Hodgkin-Lymphomen und kutanen T-Zell-Lymphomen zu ermöglichen. Für keine der beiden Studien wurden bisher Ergebnisse veröffentlicht.
Immunsuppressives Tumormilieu
In gesunden Individuen verhindern Zellen des angeborenen und des erworbenen Immunsystems (u.a. NK-Zellen, CD8+
T-Zellen) die Entstehung von Tumoren. Diese Zellen sind in der Lage, eine Immunantwort gegen maligne transformierte Zellen hervorzurufen und letztlich deren Lyse zu vermitteln. Andererseits kommt es zur Tumorgenese, wenn entartete Zellen entsprechende Immunantworten unterdrücken oder ihnen anderwärtig entgehen können.20
In etablierten soliden Tumoren herrscht in der Regel ein komplexes, immunsuppressives Milieu, welches eine effektive Aktivierung endogener und adoptiv transferierter T-Zellen unterdrückt und einen dysfunktionalen Status der T-Zellen bedingt.21, 22
Die lokale Immunsuppression innerhalb des Tumors kann unter anderem kontaktvermittelt durch sogenannte Immuncheckpoints (z.B. „programmed cell death protein 1“ [PD-1]), durch lösliche Faktoren (z.B. „transforming growth factor β“ [TGF-β]) oder durch physikalische Gegebenheiten (z.B. Hypoxie) bedingt sein.
Im Folgenden werden beispielhafte immunsuppressive Mechanismen und mögliche therapeutische Interventionen beleuchtet. Hierbei ist zu erwähnen, dass das Außer-Kraft-Setzen immunmodulatorischer Mechanismen mit dem Auftreten von schweren Nebenwirkungen (z.B. Autoimmunreaktionen) einhergehen kann.
Die immunsuppressive Funktion des inhibitorischen Rezeptors PD-1 auf CAR-T-Zellen wird durch Interaktion mit PD-L1 auf Tumorzellen oder regulatorischen Immunzellen vermittelt. Ergebnisse präklinischer Studien legen nahe, dass die Überwindung dieses Mechanismus zu einer verbesserten therapeutischen Wirksamkeit adoptiver T-Zell-Therapien führt.23
Die Kombinationstherapie von CAR-T-Zellen mit der Blockade der PD-L1/PD-1-
Achse durch Antikörper (z.B. Pembrolizumab) wird bereits in unterschiedlichen Ausführungen in klinischen Studien getestet.24
Neben der Kombinationstherapie zeigt auch der Knockout von PD-1 in CAR-T-Zellen vielversprechende präklinische Ergebnisse. Eine Arbeitsgruppe hat mittels CRISPR-Cas9 PD-1-defiziente CAR-T-Zellen entwickelt, die in einem präklinischen Tumormodell verbesserte antitumorale Wirksamkeit aufweisen.25 Ein anderer Ansatz zielt darauf ab, das inhibitorische Signal des Checkpoints in ein aktivierendes umzuwandeln. Die therapeutischen T-Zellen werden mit einem sogenannten chimären Switch-Rezeptor ausgestattet. Beispielhaft hierfür ist der PD-1-CD28-Fusionsrezeptor. Dieser vereint die extrazelluläre Domäne von PD-1 mit der transmembranen und intrazellulären Domäne von CD28. Eine Interaktion mit PD-L1 auf Tumorzellen oder regulatorischen Immunzellen resultiert dann – anstatt in einem immunsuppressiven Signal – in einem kostimulatorischen Signal.26
In etablierten soliden Tumoren modulieren verschiedene lösliche Faktoren die Eigenschaften und die Funktionalität von Leukozyten und unterdrücken somit anti-tumorale Immunantworten. TGF-β ist ein immunmodulatorisches Zytokin, welches von Tumorzellen und Tregs ausgeschüttet wird. Es inhibiert die Aktivierung, Proliferation und Differenzierung von T-Zellen.27 Die Ausstattung von CAR-T-Zellen mit einem „dominant negative TGF-β receptor 2“ schützt sie vor den immunsuppressiven Effekten von TGF-β. Dieser Rezeptor funktioniert, indem er TGF-β bindet, ohne eine intrazelluläre Signaltransduktion auszulösen, und das Zytokin somit aus der Umgebung eliminiert.28 Basierend auf vielversprechenden präklinischen Studien wird dieser Ansatz nun in einer Phase-I-klinischen Studie getestet (NCT03089203).
Regulatorische Immunzellen prägen das immunsuppressive Tumormilieu und verhindern über zahlreiche Mechanismen antitumorale Immunantworten. Das Medikament Mogamulizumab, welches CCR4+ Immunzellen (und somit Tregs) dezimiert, ist derzeit als Kombinationstherapie mit dem Anti-PD1-Antikörper Nivolumab
ineiner Phase-I/II klinischen Studie (NCT0270510) in Erprobung. Auch CAR-T-Zell-Therapien könnten von entsprechenden Behandlungsstrategien profitieren.
Mangel an spezifischen Tumorantigenen
Eine der größten Hürden in der Entwicklung klinisch einsetzbarer CAR-T-Zell-Therapien stellt die Identifikation passender Antigene dar. Chimäre Antigenrezeptoren bestehen aus einem extrazellulären, antikörperbasierten „single chain variable fragment“ (scFv), welches die Antigenspezifität bedingt, und Bestandteilen des
T-Zell-Rezeptors, welche eine intrazelluläre Signaltransduktion vermitteln.29 Dieser Aufbau ermöglicht die Erkennung von Proteinen, die auf der Oberfläche von Tumorzellen exprimiert werden.
Nur etwa 3000 Gene codieren für Oberflächenproteine, wobei auf jedem Tumor nur ein Bruchteil dieser Proteine tatsächlich exprimiert wird. Zugleich kommt so gut wie jedes dieser Proteine auch in gesundem Gewebe vor. Dieser Mangel an Tumorspezifität führt zur On-Target-Off-Tumor-Aktivierung der CAR-T-Zellen, was zu schwerer Toxizität führen kann. CD19 ist ebenfalls nicht tumorspezifisch, sondern wird auch auf allen gesunden B-Zellen exprimiert. Eine Anti-CD19-CAR-T-Zell-Therapie geht daher mit einer Depletion der gesunden B-Zellen einher. In der Hämatologie ist die Depletion von B-Zellen eine tolerierbare, weil auch führbare Nebenwirkung, die in einigen Fällen zusätzliche Maßnahmen wie die Substitution von Immunglobulinen erfordert.30
Eine Möglichkeit, die Toxizität von CAR-T-Zellen zu reduzieren, stellen Logic-Gating-Strategien dar, welche die Expression des CARs oder die CAR-T-Zell-Aktivierung an „UND“-, „ODER“- und „WENN“-Bedingungen knüpfen (z.B. Anwesenheit Antigen 1 UND Antigen 2 oder WENN Hypoxie DANN Expression des CARs). Die Funktionalität derartiger Strategien konnte in vorklinischen Studien gezeigt werden.31, 32
Um den CAR-T-Zellen einen Zugriff auf intrazelluläre Tumorantigene und somit eine viel größere Menge an möglichen Zielen zu ermöglichen, zielen Bemühungen darauf ab, scFv zu entwickeln, die „Major histocompatibility complex“(MHC)-Peptid-Komplexe erkennen.33 Ein naheliegender Ansatz ist auch die Verwendung eines
T-Zell-Rezeptors (TCR) anstelle eines CARs. Neben klassischen TCRs, die im Kontext einer MHC-Präsentation in der Lage sind, praktisch jedes Peptid zuerkennen, könnten mithilfe nichtklassischer TCRs auch andere Moleküle wie Lipide oder Riboflavin-Derivate erkannt werden.34, 35
Innerhalb solider Tumoren und zwischen verschiedenen Tumorherden herrscht außerdem eine bedeutsame Heterogenität, die Antigenexpression, Anzahl an Mutationen, Beschaffenheit der extrazellulären Matrix und Zusammensetzung der Immunzellpopulationen umfasst. CAR-T-Zellen, die lediglich Spezifität für ein Antigen aufweisen, sind deshalb möglicherweise unzureichend, um dieser Komplexität gerecht zu werden.
Neben den Herausforderungen, die solide Tumoren der neuen Therapieform stellen, limitieren weitere Aspekte die therapeutische Wirksamkeit von CAR-T-Zellen (u.a. Herstellungsprozess, Limitationen in der Gentechnik, Erschöpfung der CAR-T-Zellen). In unserem Artikel zu Faktoren, die über den Erfolg von CAR-T-Zell-Therapien bestimmen, werden diese ausführlich beleuchtet.36
Zusammenfassung
CAR-T-Zellen sind bis heute weitestgehend ineffektiv gegen solide Tumoren. Charakteristische Eigenschaften solider Tumoren, wie ein immunsuppressives Milieu und Heterogenität, stellen Hindernisse für eine erfolgreiche CAR-T-Zell-Therapie dar (Abb. 1). Damit in der Zukunft mehr Patienten von der neuen Therapieform profitieren können, müssen therapeutische T-Zellen entwickelt werden, die in den Tumor infiltrieren, der tumorassoziierten Immunsuppression entgehen und gezielt Tumorzellen erkennen und deren Lyse vermitteln. Um dies zu realisieren, bedarf es eines besseren Verständnisses der Tumor- und CAR-T-Zell-Biologie. Auch Fortschritte in der Gentechnik, die immer komplexere Modifikationen der T-Zellen ermöglichen, und neue biomathematische und bioinformatische Methoden zur Identifikation geeigneter therapeutischer Zielstrukturen könnten bei der Weiterentwicklung von CAR-T-Zellen eine wichtige Rolle spielen.
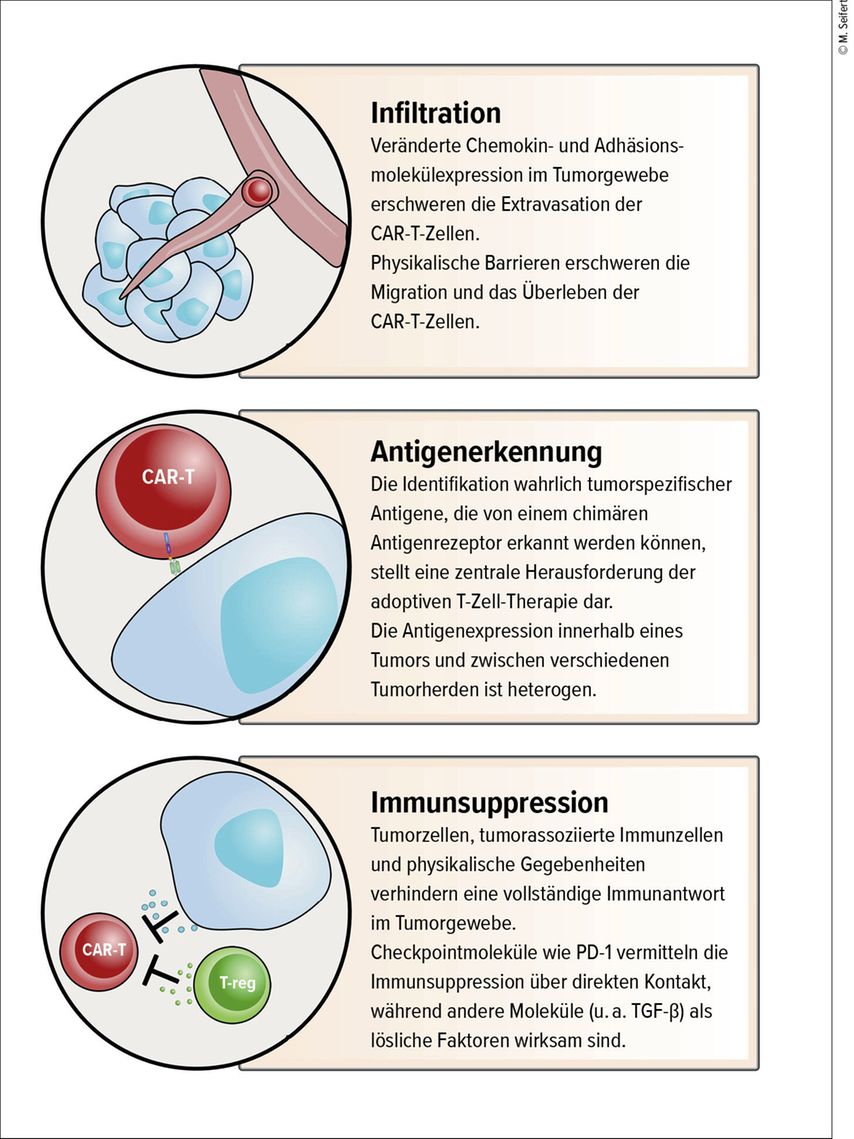
Abb. 1:Überblick über Resistenzmechanismen solider Tumoren gegen CAR-T-Zellen
Autoren:
Matthias Seifert, cand. med.1
Prof. Dr. Sebastian Kobold1–3
1 Center of Integrated Protein Science Munich (CIPS-M) und Abteilung für Klinische Pharmakologie
Medizinische Klinik und Poliklinik IV
Klinikum der Universität München
2Deutsches Konsortium für Translationale Krebsforschung (DKTK)
Standort München
3 Einheit für Klinische Pharmakologie (EKLiP) Helmholtz Zentrum München
Deutsches Forschungszentrum für Gesundheit und Umwelt (HMGU), Neuherberg
E-Mail: sebastian.kobold@med.uni-muenchen.de
Literatur:
1 Bishop MR et al.: Blood Adv 2019; 3(14): 2230-6 2 Locke FL et al.: Lancet Oncol 2019; 20(1): 31-42 3 Maude SL et al.: N Engl J Med 2018; 378(5): 439-48 4 Neelapu SS et al.: N Engl J Med 2017; 377(26): 2531-44 5 Schuster SJ et al.: N Engl J Med 2019; 380(1): 45-56 6 Ahmed N et al.: J Clin Oncol 2015; 33(15): 1688-96 7 Beatty GL et al.: Gastroenterology 2018; 155(1): 29-32 8 Galon J et al.: Science 2006; 313(5795): 1960-4 9 Kmiecik J et al.: J Neuroimmunol 2013; 264(1-2): 71-83 10 Piersma SJ et al.: Cancer Res 2007; 67(1): 354-61 11 Slaney CY et al.: Cancer Res 2014; 74(24): 7168-74 12 von Andrian UH et al.: Proc Natl Acad Sci U S A 1991; 88(17): 7538-42 13 Kadambi A et al.: Cancer Res 2001; 61(6): 2404-8 14 Lu P et al.: J Cell Biol 2012; 196(4): 395-406 15 Curiel TJ et al.: Nat Med 2004; 10(9): 942-9 16 Facciabene A et al.: Nature 2011; 475(7355): 226-30 17 Brown CE et al.: Clin Cancer Res 2015; 21(18): 4062-72 18 Caruana I et al.: Nat Med 2015; 21(5): 524-9 19 Di Stasi A et al.: Blood 2009; 113(25): 6392-402 20 Dunn GP et al.: Nat Immunol 2002; 3(11): 991-8 21 Thommen DS, Schumacher TN: Cancer Cell 2018; 33(4): 547-62 22 Rabinovich GA et al.: Annu Rev Immunol 2007; 25: 267-96 23 Iwai Y et al.: Proc Natl Acad Sci U S A 2002; 99(19): 12293-7 24 Grosser R et al.: Cancer Cell 2019; 36(5): 471-82 25 Rupp LJ et al.: Sci Rep 2017; 7(1): 737 26 Kobold S et al.: J Natl Cancer Inst 2015; 107(8): pii: djv146 27 Thomas DA, Massague J: Cancer Cell 2005; 8(5): 369-80 28 Kloss CC et al.: Mol Ther 2018; 26(7): 1855-66 29 Guedan S et al.: Mol Ther Methods Clin Dev 2019; 12: 145-56 30 Yamamoto TN et al.: Nat Med 2019; 25(10): 1488-99 31 Juillerat A et al.: Sci Rep 2017; 7: 39833 32 Srivastava S et al.: Cancer Cell 2019; 35(3): 489-503 e8 33 Maus MV et al.: Mol Ther Oncolytics 2016; 3: 1-9 34 Crowther MD et al.: Nat Immunol 2020; 21(2): 178-85 35 Vavassori S et al.: Nat Immunol 2013; 14(9): 908-16 36 Lesch S et al.: Semin Cancer Biol 2019; pii: S1044-579X(19)30219-6 (E-pub ahead of print)
Das könnte Sie auch interessieren:

«Können für das Management von Bedeutung sein»
Modic Changes beschreiben Veränderungen in der Wirbelsäulen-MRT, die auf den Radiologen Dr. Michael Modic zurückgehen. Welchen Stellenwert die Veränderungen haben und wann sie ...
Nur eine von vielen Begleiterscheinungen
Warum er MRT-Veränderungen bei unspezifischen Rückenschmerzen wenig Bedeutung beimisst, erklärt Prof. Dr. med. Andreas Seekamp aus Kiel.
Modic Changes bei Rückenschmerzen
Obwohl die Erstbeschreibung fast 40 Jahre her ist, ist immer noch nicht klar, wodurch die MRT-Veränderungen entstehen und welchen Stellenwert sie für Diagnostik und Therapie haben.