Quo vaditis, myelodysplastische Syndrome/myelodysplastische Neoplasien?
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Zwei aktualisierte Klassifikationen zu Lymphomen und myeloischen Neoplasien umfassen zwar thematisch ähnliche Gefilde, unterscheiden sich aber in einigen relevanten Punkten.
Wahrnehmbar und objektiv belegbar ist in unserer komplexen Welt eine Zunahme der Polarisierung festzustellen.1 Diese hat auch vor der Academia keinen Halt gemacht. Nur 28 Jahre nach den beispielhaften internationalen Bemühungen, alle Differenzen in den Klassifikationen der hämatolymphoiden Tumoren auszuräumen, die in der Entstehung der universell anerkannten dritten, vierten und der revidierten vierten Auflage der WHO-Klassifikation mündeten, haben in den Jahren 2021–22 Zentrifugalkräfte in der Gemeinschaft der Hämatopatholog:innen und Hämatoonkolog:innen die Oberhand gewonnen.
Das Ergebnis war die Entwicklung von zwei unabhängigen Klassifikationen sowohl der Lymphome als auch der myeloischen Neoplasien, die kurz aufeinanderfolgend im Sommer 2022 in den renommierten Journalen Blood2 und Leukemia3 publiziert wurden: die Klassifikationen ICC 2022 und WHO 5.
Lost in Translation: MDS
Zwar lassen sich beide Klassifikationen weitestgehend ineinander übersetzen (Tab. 1),4 dennoch wird das myelodysplastische Syndrom (MDS) in den Klassifikationen mit zwei unterschiedlichen Namen geführt, nämlich in ICC 20222 als MDS und in WHO 5 als myelodysplastische Neoplasie, die aber immer noch traditionell mit MDS (und nicht mit MDN) abgekürzt wird.3 Auch werden einige Subentitäten von der jeweils anderen Klassifikation nicht geführt und die Kriterien für die Klassifikation einiger Subentitäten weisen deutliche Unterschiede auf.
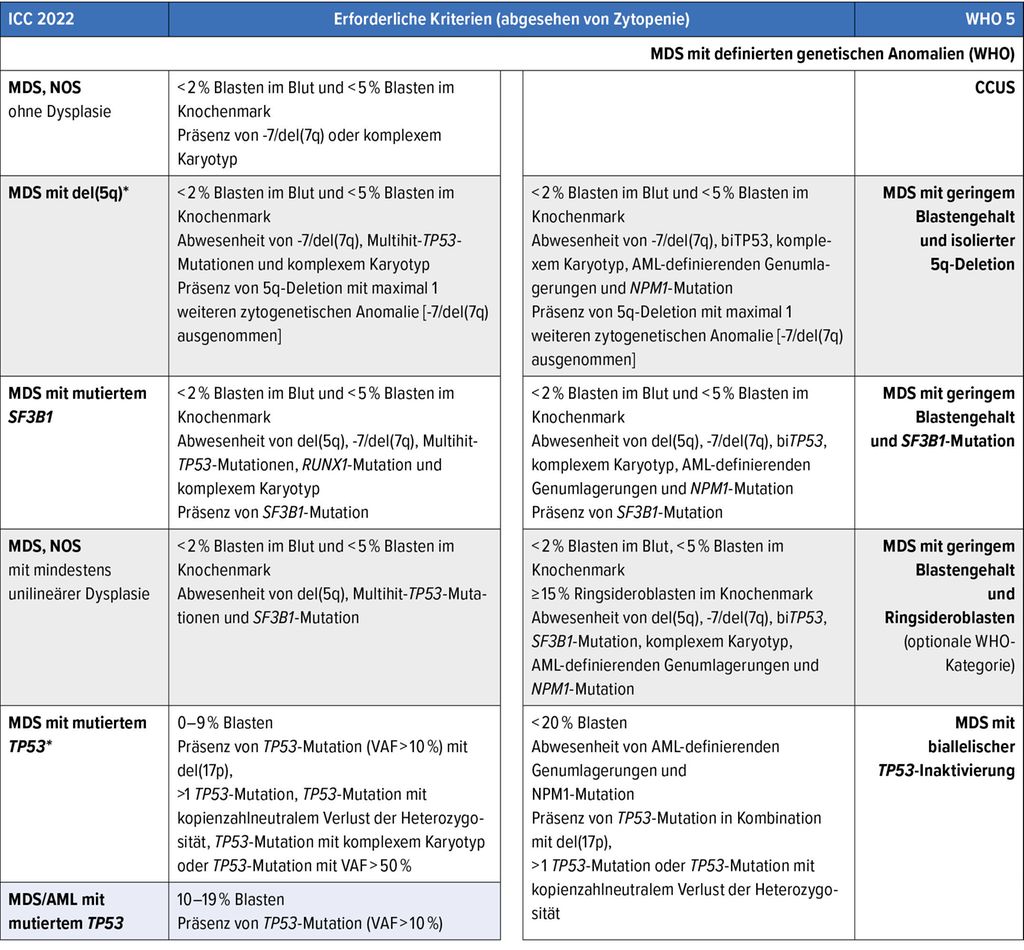
Tab. 1: Gegenüberstellung der Klassifikation der myelodysplastischen Syndrome (MDS)/myelodysplastischen Neoplasien: Internationale Konsensklassifikation 2022 (ICC 2022) vs. 5. Weltgesundheitsorganisations-Klassifikation (WHO 5); farbig hinterlegte Kategorien fehlen in der jeweils anderen Klassifikation
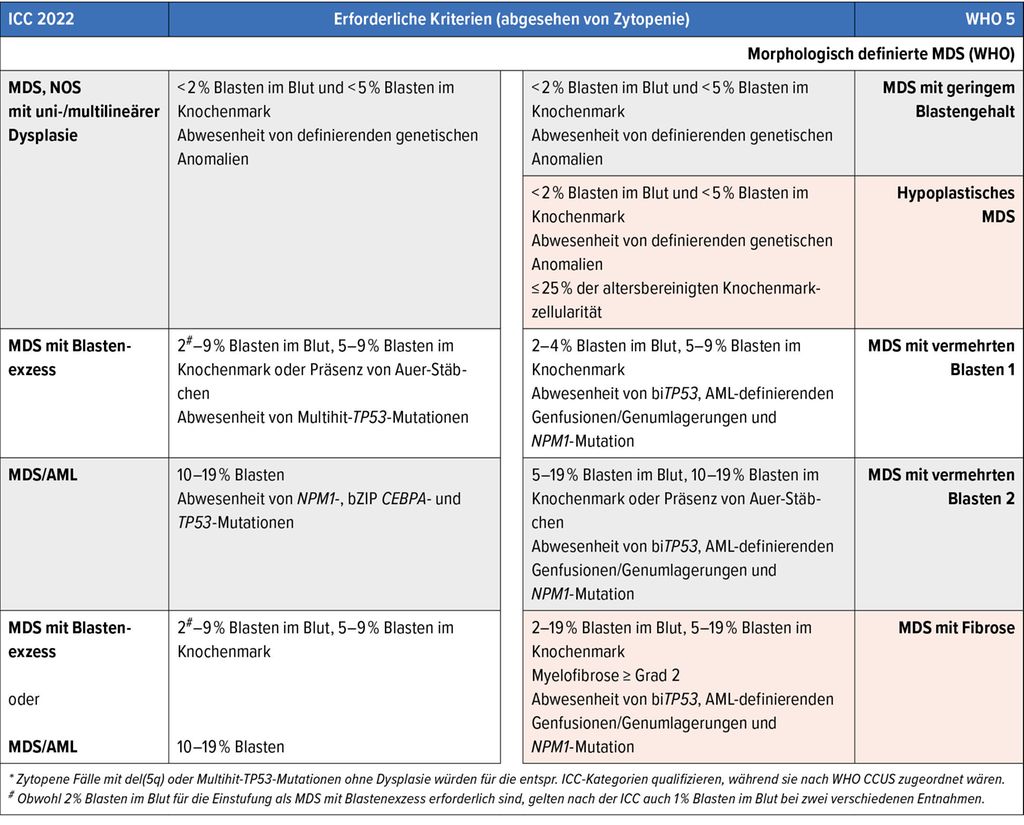
Tab. 1: (Fortsetzung)
Wenn man beide Positionspapiere zu den Klassifikationen2,3 studiert, so zeigt sich, dass die wissenschaftliche Evidenz für die Neuauflage der revidierten WHO 4, auf der beide Klassifikationen, ICC 2022 und WHO 5, aufbauen, zwar fast identisch war, jedoch wurde diese unterschiedlich interpretiert.
Die ICC lässt aufgrund der potenziellen Subjektivität der Erhebung der Präsenz von Dysplasie (Abb. 1A&B) und der exakten Blastenmenge5 weniger Gewicht zukommen, legt dafür aber mehr Wert auf die zugrunde liegenden wiederkehrenden genetischen Anomalien. Die WHO-Klassifikation folgt mehr dem traditionellen MDS-Klassifikationsmodell.6
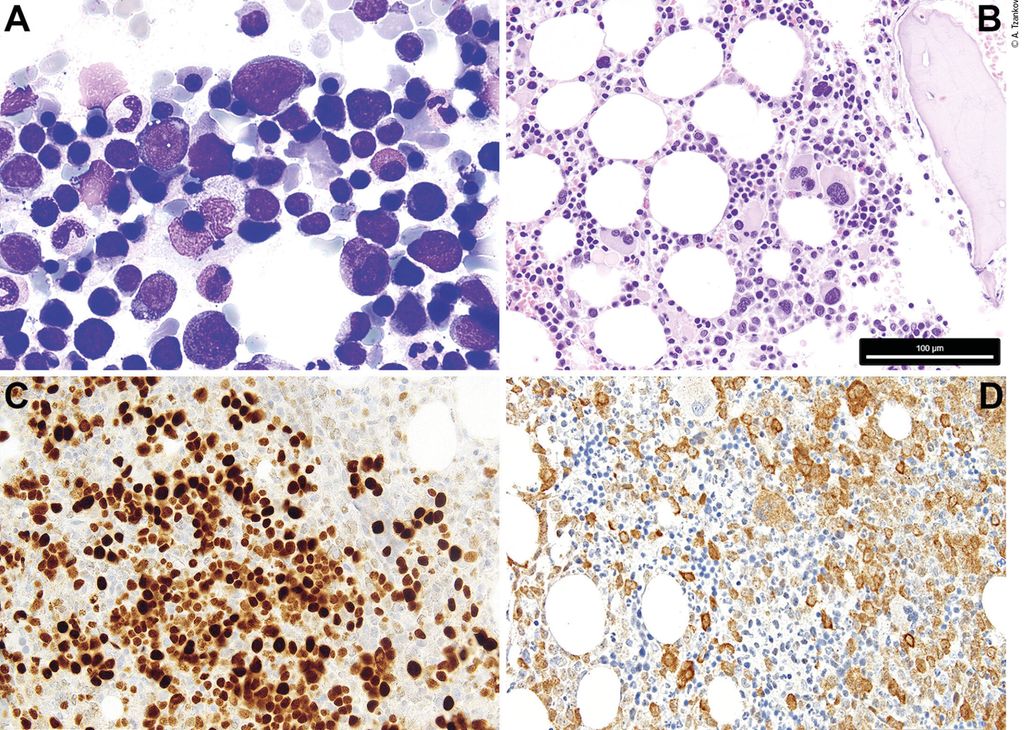
Abb. 1: 1A: Zytologie eines MDS mit Blastenexzess/vermehrten Blasten; man beachte die kleinen dysplastischen Megakaryozyten, die Reifungsstörung und die Granulationsdefekte der Myelopoese, die spärliche Dyserythropoese und 7% Blasten. 1B: Histopathologie eines MDS mit del(5q) und monoallelischer TP53-Mutation; man beachte das Vorhandensein von einigen nukleär monolobierten, kleineren Megakaryozyten, nebst Megakaryozyten mit separierten Kernen, und die myelopoetische Reifungsstörung. 1C: charakteristische immunhistochemische p53-Positivität bei MDS mit Multihit-TP53-Mutationen/biallelischer TP53-Inaktivierung (6% Blasten); das mutierte p53-Protein verfügt über eine längere Halbwertszeit und stabilisiert allfällig (noch) vorhandenes Wildtyp-p53, sodass sich das entsprechende Protein, das ansonsten im Knochenmark kaum immunhistochemisch nachweisbar ist, im Falle einer signifikanten mutierten Allellast wie bei Multihit-TP53-Mutationen/biallelischer TP53-Inaktivierung färberisch darstellen lässt. 1D: MDS mit Blastenexzess (8% Blasten) und Positivität mit dem mutationsspezifischen Anti-NPM1-Antikörper (PA1-46356); nach WHO muss der Fall als AML klassifiziert werden, während nach ICC die Diagnose MDS mit Blastenexzess zulässig ist. Der Messbalken in B bezieht sich auf die Abb. B–D
Dies zeigt sich auch in der MDS-Definition: Zwar ist die Präsenz mindestens einer nicht anderwärtig zu erklärenden Zytopenie (i.e. Anämie mit Hämoglobinwerten <120g/l für Frauen und <130g/l für Männer, Neutropenie von <1,8x109/l und/oder Thrombopenie von <150x109/l) in beiden Klassifikationen identisch, jedoch wird bei Vorhandensein der MDS-definierenden genetischen Anomalien – del(5q), Multi-hit-TP53-Mutationen, SF3B1Mutationen, -7/del(7q) oder komplexer Karyotyp – die Präsenz von Dysplasie von der ICC für die Diagnose nicht mehr gefordert und die entsprechenden Fälle werden als «MDSohne morphologischeDysplasie» klassifiziert. Die WHO hingegen klassifiziert mit Ausnahme von «MDSmit geringer Blastenmenge» und «isolierter5q-Deletion» allfällige dysplasiefreie Fälle als klonale Zytopenien ungewisser Signifikanz (CCUS).
In dieser Hinsicht liess die ICC der starken, in diesem Fall positiven prognostischen und auch prädiktiven Bedeutung von del(5q) (was Lenalidomid-Sensitivität anbelangt), aber vor allem der negativen prognostischen Bedeutung der anderen aufgezählten genetischen Anomalien7,8 ein diagnostisches Gewicht zukommen, während die WHO dies nur bei del(5q) zulässt. Daraus folgen ebenfalls Übersetzungsschwierigkeiten (Tab. 1).
Mit dieser Ausnahme fordern allerdings beide Klassifikationen für die Abgrenzung von MDS gegenüber CCUS in allen anderen Fällen die Identifikation von >10% Dysplasien in den hämatopoetischen Reifungsreihen.
Abgrenzungsschwierigkeiten: AML
Weitere Abgrenzungsschwierigkeiten ergeben sich hinsichtlich akuter myeloischer Leukämien (AML). Hier zieht das Vorhandensein von AML-definierenden genetischen Anomalien, mit wenigen Ausnahmen unabhängig von der Blastenmenge, die WHO-Diagnose einer AML3 mit sich. Nach der ICC-Klassifikation2 ist dies erst ab einer Blastenmenge von >10% der Fall. So ist z.B. ein MDS mit NPM1-Mutation nach WHO keine zulässige Diagnose, während dies nach ICC bis zu einer Blastenmenge von <10% zulässig ist (Abb. 1D).
Gerade in der Abgrenzung AML gegenüber MDS ergibt sich ein weiterer relevanter Unterschied zwischen beiden Klassifikationen: Die ICC hat die Entität «MDS/AML» für Fälle mit 10–19% Blasten im peripheren Blut und/oder im Knochenmark bei Fehlen von AML-definierenden genetischen Anomalien ausgerufen (gilt nicht für pädiatrische MDS [<18 Jahre]).2 Die Schaffung dieser Kategorie wird dadurch gestützt, dass die Prognose von Patient:innen mit oligoblastischer AML (20–30% Blasten) und solchen mit der WHO-Kategorie der «MDS mit vermehrten Blasten2» (die von der ICC nicht mehr geführt wird) vergleichbar ist.5 Der praktische Vorteil ist, dass die neue MDS/AML-Kategorie die Aufnahme von Patient:innen sowohl in MDS- als auch AML-Studien formalisiert und so Verhandlungen mit Kostenträgern erleichtern kann, was zugegebenermassen durch die WHO ebenfalls diskutiert wird,3 aber dennoch in die Klassifikation nicht einfliesst.
Ringsideroblasten ade?
Die ICC-Kategorie «MDSmit mutiertem SF3B1»/WHO-Kategorie «MDS mit geringer Blastenmenge und SF3B1-Mutation» ersetzen die meisten Fälle der alten WHO-4-Kategorie «MDSmit Ringsideroblasten» (MDS-RS). Die neue ICC-Klassifikation verzichtet vollständig auf MDS-RS: Fälle mit RS, bei denen eine SF3B1-Mutation fehlt, werden als «MDS, NOS» klassifiziert, während sie von der WHO optional als «MDS mit geringer Blastenmengeund RS» (sofern diese ≥15% ausmachen) eingestuft werden.
Die ICC-Rationale dahinter ist, dass die Biologie und die günstige Prognose der MDS-Fälle mit RS und SF3B1-Mutation durch das Vorhandensein der Mutation [wenn – durch die ICC expliziert erwähnt2 – nicht in Begleitung von del(5q), ungünstigem Karyotyp, RUNX1- oder TP53-Mutationen] und nicht durch die Anwesenheit von RS bestimmt werden.9 Dennoch rät auch die ICC, nach RS aktiv zu suchen, weil sie Ausdruck einer Dyserythropoese sind und somit zur Diagnose von MDS beitragen und ihr Vorhandensein eine allfällige SF3B1-Mutation und möglicherweise ein Ansprechen auf Luspatercept10 voraussagt.
TP53-Mutationen
Beide Klassifikationen messen TP53-Mutationen8,11 eine grosse Bedeutung bei und führen neu die Kategorien der «MDS mit multihit TP53-Mutationen» (0–9% Blasten) und «MDS/AMLmit mutiertem TP53» (10–19% Blasten; ICC) bzw. «MDS mit biallelischer TP53-Inaktivierung» (biTP53; 0–19% Blasten; WHO) ein, die sämtliche andere MDS-Kategorien übertrumpfen. Multihit bzw. biTP53 wird definiert durch TP53-Mutation in Kombination mit del(17p), >1 TP53-Mutation oder TP53-Mutation mit kopienzahlneutralem Verlust der Heterozygosität,2,3 was sich oft in einer Variantenallelfrequenz (VAF) von >50% widerspiegelt. Die ICC lässt in dieser Hinsicht auch TP53-Mutationen in Kombination mit komplexerem Karyotyp zu und fordert explizit eine VAF von ≥10% für die TP53-Mutationen.2
Ein sehr wesentlicher Unterschied zwischen beiden Klassifikationen ergibt sich in Fällen mit 10–19% Blasten: In solchen genügt nach ICC, wie es auch die Literaturevidenz suggeriert,11 das Vorhandensein einer TP53-Mutation für die Diagnose MDS/AML mit mutiertem TP53,2 während nach WHO ein biTP53-Status für die Diagnose MDS-biTP533 vorliegen muss.
ICC-Manko: hypoplastisches MDS
Das hypoplastische MDS (MDS-h) wird ausschliesslich von der WHO geführt und wird definiert als MDS ohne definierende genetische Anomalien mit <5% Blasten und ≤25% der altersbereinigten Knochenmarkzellularität.3 Das MDS-h ist für seine charakteristischen Merkmale,12 insbesondere potenzielles Ansprechen auf Immunsuppression,13 bekannt und scheint auch ein spezifisches Profil mit weniger Splicing-Faktor-kodierenden und mehr RNA-Helicase-kodierenden Genmutationen aufzuweisen,14 sodass das Nichtaufführen durch die ICC ein Manko darstellt.
Dies scheint bei der zweiten Kategorie, die von der ICC nicht geführt wird, nämlich bei «MDS mit Fibrose»,3 eher nicht der Fall zu sein: Die Fibrose ist bei MDS zwar ein blastenzahlunabhängiger prognostischer Parameter,15 allerdings ist sie eng mit Alter, Blastenzahl, WHO-4-MDS-Kategorie, TP53- und SETBP1-Mutationen und prognostisch ungünstigem Karyotyp verbunden16 und ein einheitlicher Schwellenwert, Fibrosegrad ≥2 oder ≥3,15,16wird nicht allgemein anerkannt, sodass unklar ist, ob der Myelofibrosegrad ≥2 bei MDS-Fällen mit 5–19% Knochenmarkblasten Ausdruck einer spezifischen Biologie ist, so wie die WHO es suggeriert,3 oder ein einprägsames Epiphänomen darstellt.
Fazit
Der Komplexitätsgrad der MDS-Dia-gnostik ist mit der Einführung zweier Klassifikationen im Jahr 2022 gestiegen. Neben klinischen Informationen, die das Mindesterfordernis darstellen (Differenzialblutbild, Anamnese hinsichtlich zytotoxischer Therapie, Familienanamnese hinsichtlich Keimbahnprädisposition), sind Laborprüfergebnisse (Dyshämatopoese und Blastenmengen im Knochenmark, Karyotypisierung für MDS-spezifische Anomalien bzw. zum Ausschluss AML-spezifischer Anomalien und Mutationsuntersuchungen zumindest für SF3B1, TP5317, NPM118 und CEBPA; Abb. 1C&D) und auch eine Übersetzung der spezifischen MDS-Subkategorien erforderlich, vom ICC- in den WHO-Wortlaut und umgekehrt.
Literatur:
1 Ross Arguedas A et al.: Oxford 2022: Reuters Institute, University of Oxford. Online unter https://reutersinstitute.politics.ox.ac.uk/echo-chambers-filter-bubbles-and-polarisation-literature-review. Abgerufen am 29.12.2023 2 Arber DA et al.: Blood 2022; 140(11): 1200-28 3 Khoury JD et al.: Leukemia 2022; 36(7): 1703-19 4 Zeidan AM et al.: Leukemia 2022; 36(12): 2939-46 5 Estey E et al.: Blood 2022; 139(3): 323-32 6 Bennett JM et al.: Br J Haematol 1982; 51(2): 189-99 7 Bernard E et al.: Nat Med 2020; 26(10): 1549-56 8 Grob T et al.: Blood 2022; 139(15): 2347-54 9 Malcovati L et al.: Blood 2020; 136(2): 157-70 10 Amitai I et al.: Blood 2023; 142(Suppl. 1): 6487 11 Weinberg OK et al.: Blood Adv 2022; 6(9): 2847-53 12 Bono E et al.: Leukemia 2019; 33(10): 2495-505 13 Passweg JR et al.: J Clin Oncol 2011; 29(3): 303-9 14 Nazha A et al.: Haematologica 2015; 100(11): e434-7 15 Della Porta MG et al.: J Clin Oncol 2009; 27(5): 754-62 16 Melody M et al.: Clin Lymphoma Myeloma Leuk 2020; 20(5): 324-8 17 McGraw KL et al.: Haematologica 2016; 101(8): e320-3 18 Patel SS et al.: Mod Pathol 2020; 33(7): 1380-8
Das könnte Sie auch interessieren:
SOC in Diagnose und Management
Unter dem Vorsitz von Prim. Priv.-Doz. Dr. Birgit Volgger, Bezirkskrankenhaus Lienz, und Univ.-Prof. Dr. Christian Marth, MedUni Innsbruck, fand am 10. Mai 2025 im Rahmen der ...
Hautmanifestationen bei onkologischen Erkrankungen
Krebserkrankungen verschiedener Organsysteme können auch mit Symptomen an der Haut einhergehen, die manchmal bereits als frühe Warnzeichen auftreten. Dazu zählt ausgeprägter Pruritus. ...
Therapie des multiplen Myeloms
Dank vielfältiger neuer Therapieoptionen verbessert sich die Prognose beim multiplen Myelom (MM) stetig. Die bereits verfügbaren Therapien können oft eine langfristige, zum Teil auch ...