Sarkome: selten, aber aggressiv
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Das aktuelle Cancer Update des Comprehensive Cancer Center Vienna beleuchtete das Thema Weichteiltumoren und Sarkome aus verschiedenen Blickwinkeln von der Diagnostik bis zur Therapie. Durch die Veranstaltung führten Univ.-Prof. Dr. Reinhard Windhager, Universitätsklinik für Orthopädie und Unfallchirurgie der MedUni Wien/AKH Wien, und Univ.-Prof. Dr. Thomas Brodowicz, Universitätsklinik für Innere Medizin I der MedUni Wien/AKH Wien.
In seiner Begrüßung erklärte Prof. Reinhard Windhager, dass Sarkome nur etwa 1% der Tumoren ausmachen. Wegen ihrer Aggressivität seien sie aber berüchtigt und für die Betroffenen eine enorme Gefahr.1
Zeit für das Bild – die Bildgebung
Univ.-Prof. PD Dr. Iris Nöbauer, Expertin für muskuloskelettale Radiologie an der Universitätsklinik für Radiologie und Nuklearmedizin, erläuterte das diagnostische Vorgehen. Nach Erheben der Anamnese und Begutachten eventuell bereits vorhandener Aufnahmen wird bei der klinischen Untersuchung beurteilt, ob die Läsion palpabel, hart oder weich ist, ob sie verschieblich und/oder schmerzhaft ist, ob es sich um eine einzelne oder um multiple Läsionen handelt. Außerdem wird auf Hautveränderungen wie eine pathologische Gefäßzeichnung geachtet. Ist die Veränderung zugänglich und kleiner als fünf Zentimeter, dann sei die Ultraschalluntersuchung die Methode der Wahl, sagte Nöbauer.2 So könnten bereits viele benigne Läsionen charakterisiert werden, etwa Zysten, oberflächliche Lipome, Fremdkörpergranulome etc. Sie warnte davor, klar begrenzte Läsionen immer als benigne einzustufen: „Eine glatte Begrenzung ist beim Weichteiltumor kein Zeichen für Gutartigkeit!“ Im Zweifel sollte eine MRT erfolgen.2
Bei allen Veränderungen, die größer als fünf Zentimeter oder sonografisch suspekt sind, ist eine MRT angezeigt – und zwar VOR einer Biopsie, nicht danach, wie Nöbauer betonte. Die MRT ist immer indiziert bei tiefen und schnell wachsenden Läsionen mit Knochen- oder Gelenkbeteiligung.2 Bei allen Veränderungen, die auch mittels MRT nicht charakterisiert werden können, muss eine Biopsie entnommen werden, wobei eine bildgesteuerte Biopsie die Methode der Wahl ist. „Wichtig ist, dass man vitales Tumorgewebe und genügend Proben hat“, sagte Nöbauer. Deshalb sollte die Biopsie in einem Sarkom-Referenzzentrum entnommen und der Zugang in Absprache mit den Operateur:innen gewählt werden.2
Bei Sarkomen ist generell ein Ganzkörper-Staging nötig. Die Methode der Wahl ist dabei eine Thorax-CT. Bei Verdacht auf Lymphknotenmetastasen ist eine Kontrastmittel-gestützte CT angeraten, und zum Aufspüren von Skelettmetastasen eine MRT.2 Bei PET-aviden Tumoren und entsprechender Erfahrung kann auch eine PET/CT wertvolle Hinweise liefern.
„Fast tracking“ mittels Telemedizin
Nicht immer ist ein Sarkomzentrum erreichbar. Gerade in abgelegenen Gegenden oder in besonderen Situationen wie während der Covid-19-Pandemie ist vielen Menschen der Zugang zu solchen Einrichtungen nicht möglich. Hier bietet die Telemedizin eine Alternative. Darauf ging Ap. Prof. PD Dr. Gerhard Hobusch, MSc, Universitätsklinik für Orthopädie und Unfallchirurgie, ein. „Kliniken, die keine Spezialisierung in Richtung Sarkombehandlung aufweisen, ist es heutzutage nicht möglich maligne von benignen Tumoren zu unterscheiden“, sagte er.
Während der Pandemie wurde in der Spezialambulanz für Knochen- und Weichteilsarkome am AKH Wien eine telemedizinische Tumorambulanz eingerichtet. Diese erhält vorab die entsprechenden Aufnahmen und entscheidet, ob es sich um eine maligne Läsion handelt und die Patient:innen in die Ambulanz kommen müssen oder ob es sich um eine benigne Veränderung handelt, die andernorts behandelt werden kann. Vorteile sind laut Hobusch, dass weniger Patient:innen die Ambulanz aufsuchen müssen, was den Betroffenen Zeit spart. Gleichzeitig steht mehr Zeit für die Diagnostik und Therapie der tatsächlichen Sarkompatient:innen zur Verfügung. Zunehmend von Bedeutung seien das Telecoaching und die fachgerechte Anleitung von Zuweiser:innen in der Metastasentherapie, erklärte Hobusch.
Pathologische Diagnostik
Dr. Marita Kölz, Klinisches Institut für Pathologie, führte in die pathologische Diagnostik von Sarkomen ein, die eine spezifische Expertise verlangt. Die Gründe dafür sind vielfältig. So sind die Tumoren mit 11000 Neuerkrankungen/Jahr in ganz Europa selten. Zudem sind mehr als 100 Entitäten beschrieben, die sich durch morphologische Heterogenität auszeichnen, und die Dignitätsbeurteilung weicht von jener der Karzinome ab. Bei Sarkomen gibt es neben benigne und maligne die Kategorie „intermediär“, die unterteilt wird in lokal aggressiv (z.B. Fibromatose) und selten metastasierend (z.B. solitär fibröse Tumoren). Die Dignitätszuordnung ist meist erst nach Bestimmung der Entität möglich, die auf der WHO-Klassifikation von 2020 basiert. Die Entitätszuordnung erfolgt durch Histomorphologie, Immunhistochemie und Molekularpathologie/Genetik.1Bei einem Teil der Sarkome erfolgt ein histomorphologisches Grading nach FNCLCC. Eine Befundung in einem entsprechenden Referenzzentrum ist besonders wichtig, betonte Kölz.
Für die pathologische Untersuchung sind am besten mehrere bildgesteuerte kompakte Nadelstanzbiopsien (mindestens 14G) geeignet, die aus unterschiedlichen Tumorarealen entnommen werden sollen. Inzisionsbiospien sind wegen des Auftretens von Artefakten und da hierbei nur regionale Abschnitte des Tumors erfasst werden für die Diagnostik weniger geeignet. Korrekt ausgeführt und in einem entsprechenden Referenzzentrum beurteilt, führt die Biopsie in mehr als 90% der Fälle zur richtigen Diagnose.
Sarkomchirurgie: bitte ohne „whoops“!
Prof. Reinhard Windhager beantwortete in seinem Vortrag die Frage, wie Sarkome chirurgisch behandelt werden müssen. Auf jeden Fall sei ein „Whoops“-Eingriff zu vermeiden, wenn in der Sarkomchirurgie unerfahrene Operateur:innen eine banale Läsion, etwa ein Hämatom oder Ganglien, erwarten und nach Beginn des Eingriffs feststellen, dass es sich um einen malignen Tumor handelt. Dies mache eine komplexere Operation notwendig und verschlechtere die Prognose.3 Damit zeigte Windhager auch das größte Problem auf: ungeplante Operationen von Weichteilsarkomen, die 19 bis 53% der Fälle ausmachen. Sie gehen mit einer größeren Menge an Resttumorgewebe einher und die Rate für Lokalrezidive steigt auf bis zu 67%.4
Grundlage der Sarkomchirurgie ist die Klassifikation des amerikanischen Orthopäden und Onkologen William F. Enneking. Er definierte das Grading (I–III) ebenso wie die Operationsränder (intraläsional, marginal, weit, radikal).5 Radikal bedeutete zunächst, dass das komplette tumortragende Kompartiment entfernt wurde, das heißt der ganze Muskel oder Knochen. Dies sollte verhindern, dass sogenannte Skip-Läsionen übersehen wurden, die dann zum erneuten Tumorwachstum führen. Durch verfeinerte Diagnostikmethoden konnten solche Läsionen besser dargestellt und chirurgisch entfernt werden. Derzeit würden Abstände zu gesundem Gewebe von 1mm6,7 – teilweise sogar unter 1mm8–10 als ausreichend angesehen, sagte Windhager. Voraussetzung seien eine adäquate Diagnostik, korrektes Staging/Grading und dass keine Skip-Läsionen übersehen werden (Abb. 1). Die Ergebnisse seien vor allem vom Grading abhängig: Bei gut differenzierten Tumoren liege die Überlebensrate bei etwa 85%, bei wenig differenzierten bei circa 20%, so Windhager.11 Dabei sei wichtig, dass die Patient:innen in spezialisierten Tumorzentren operiert werden, betonte er. Doch selbst in den USA werden die wenigsten Sarkompatient:innen in solchen Zentren behandelt, wie aus der größten Sarkomdatenbank hervorgeht.11
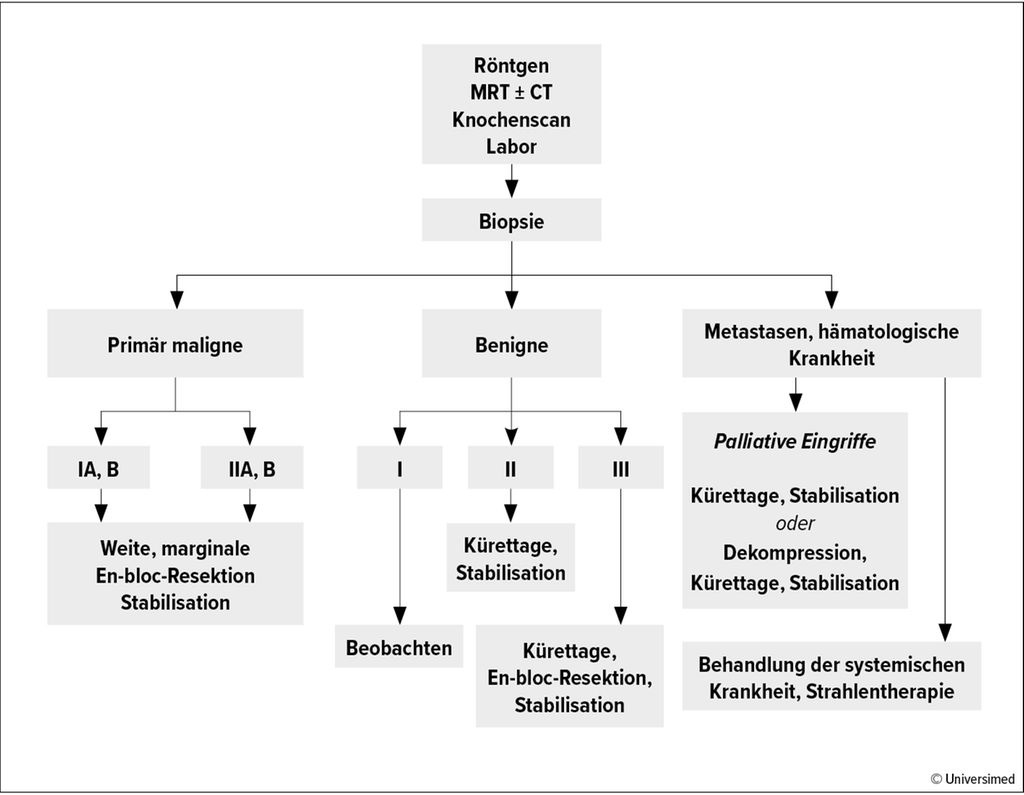
Abb. 1: Präoperativer Algorithmus für muskuloskelettale Tumoren
Tumoren des Retroperitoneums
Univ.-Prof. Dr. Anton Stift, Abteilung für Viszeralchirurgie der Universitätsklinik für Allgemeinchirurgie, beleuchtete die Tumoren des Retroperitoneums. Sie haben eine Inzidenz von 0,31–1/100000. In Österreich sind dies 90 bis 100 pro Jahr mit einem Altersgipfel von 59 bis 61 Jahren. Insgesamt sind mehr als 100 histologische Subtypen beschrieben. Am häufigsten kommen Liposarkome vor, gefolgt von Leiomyosarkomen.
Retroperitoneale Tumoren zeigen keine tumorspezifischen Symptome. Betroffene bemerken eine Veränderung ihres körperlichen Erscheinungsbildes in Form von einseitigen Vorwölbungen im Flankenbereich. Auch Verdrängungssymptome wie Unwohlsein etc. können auftreten, weshalb sie in der Regel zuerst ihre Hausärzt:innen aufsuchen. Oft sind die Tumoren auch ein Zufallsbefund im Rahmen einer Nieren- oder Abdomensonografie. Allgemeinärzt:innen sollten diese Patient:innen zur weiteren Abklärung in die Radiologie überweisen. Die Methode der Wahl ist die MRT, bei Verdacht auf eine Metastasierung auch die CT. Das weitere Vorgehen wird dann im Tumorboard besprochen. Dort wird auch geklärt, ob eine präoperative Therapie sinnvoll sein könnte, um die vollständige Resektion des Tumors zu ermöglichen. Stift wies darauf hin, dass dies erst durch die verbesserte Typisierung der Tumoren möglich wurde. Das Operationsziel ist eine En-bloc-Resektion, wobei in etwa 50% der Fälle auch benachbarte Organe mit entfernt werden müssen, wie Stift erklärte. Meist handelt es sich dabei um die Niere, sofern die kontralaterale Niere noch funktionsfähig ist, aber auch Darm, Milz, Pankreas, Gefäße und Teile der Leber. Solche Eingriffe gehen einher mit einer hohen perioperativen Morbidität von bis zu 40% und einer perioperativen Mortalität innerhalb der ersten 90 Tage post OP von 0,3–6,4%.12 Patient:innen mit retroperitonealen Tumoren gehören in ein spezialisiertes Zentrum!
Bestrahlung vorher oder nachher?
PD Dr. Franziska Eckert, Universitätsklinik für Radiologie, warf zunächst einen Blick zurück. In früheren Jahren war die Amputation die einzige Therapieoption bei Weichteilsarkomen der Extremitäten. Später wurde extremitätenerhaltend operiert und postoperativ bestrahlt. Inzwischen besteht die Möglichkeit, präoperativ mit 50 Gray (Gy) zu bestrahlen. Bei einer Dosis von 2Gy/Tag dauert die Behandlung fünf Wochen. Im Vergleich mit der postoperativen Bestrahlung (50Gy+16Gy Boost direkt ins OP-Areal) zeigen sich die gleichen onkologischen Ergebnisse. Bei der präoperativen Bestrahlung komme es aber seltener zu Spättoxizitäten wie Fibrosen oder Funktionseinschränkungen der Gelenke, bei der postoperativen dagegen zu weniger Wundheilungsstörungen, erklärte Eckert.
Bei geriatrischen Patienten ist präoperativ auch eine hypofraktionierte Strahlentherapie (25Gy in 5 Fraktionen) möglich.13 Eine präoperative Strahlentherapie ist unter anderem zu erwägen, wenn die postoperative Bestrahlung nicht möglich ist, zum Beispiel wegen der Nähe zu Organen bei retroperitonealen Tumoren. Außerdem etwa bei Tumoren in Gelenknähe, wenn Langzeitkomplikationen im Vordergrund stehen oder mit reduzierter Dosis bei myxoiden Liposarkomen.
Indikationen für eine postoperative Strahlentherapie seien exulzerierte Tumoren, unkontrollierbare Schmerzen und wenn eine problematische Wundheilung zu erwarten ist, sagte die Radioonkologin. Ihr Fazit: Die Therapie gehört an ein spezialisiertes Zentrum. Der interdisziplinäre Austausch sei wichtig, denn „Strahlentherapie ist Millimeterarbeit“. In die Planung fließen alle vorhandenen Informationen aus Bildgebung, Histologie, OP-Bericht etc. ein.
Überlebensvorteil durch systemische Therapie?
Prof. Dr. Thomas Brodowicz erklärte, dass es bei Weichteilsarkomen auf eine genaue Patient:innenselektion ankommt, um einen Überlebensvorteil durch die systemische Therapie zu erreichen. So zeigte die Subgruppenanalyse der Studie EORTC-STBSG 6293, dass nur Hochrisiko-Sarkompatient:innen einen Überlebensvorteil durch eine adjuvante Chemotherapie haben.14 Das Gleiche gilt im metastasierten Stadium, wie die Phase-III-Studie LMS-04 an Patient:innen mit metastasierten Leiomyosarkomen belegte.15 Eine Studie, die Eribulin mit Dacarbazin bei Patient:innen mit Leiomyo- und Liposarkomen verglich, ergab einen Überlebensvorteil für Eribulin und darüber hinaus, dass dieser bei Liposarkomen größer war als bei Leiomyosarkomen (15,6 vs. 8,4 Mo.).16
Bei den gastrointestinalen Stromatumoren (GIST) sind Tyrosinkinase-Inhibitoren die Therapie der Wahl in der Erstlinie wie auch in den folgenden Linien. Die Abfolge: Imatinib, dann Sunitinib, Regorafenib und zuletzt Ripretinib.
Weichteilsarkome im Kindesalter
Univ.-Prof. Dr. Leo Kager, Leiter der hämatologischen und onkologischen Ambulanz am St. Anna Kinderspital, Wien, ging auf die besonderen Herausforderungen bei Weichteilsarkomen im Kindesalter ein. Hier handelt es sich bei hochmalignen Tumoren meist um Rhabdomyosarkome. In Österreich treten etwa zehn Neuerkrankungen pro Jahr auf. Klinisch zeichnen sie sich durch eine länger bestehende schmerzlose Schwellung aus, die oft fehlinterpretiert wird. Bei Hochrisiko-Rhabdomyosarkomen im fortgeschrittenen Stadium liegt das Langzeitüberleben bei lediglich 16%.17 Glücklicherweise gehörten die meisten Patient:innen der Gruppe mit intermediärem Risiko an, bei der das Langzeitüberleben zwischen 60 und mehr als 80% liege, sagte Kager.
Für die Diagnostik gelte das ALARA-Prinzip: As Low As Reasonable Achievable – so wenig wie nötig, aber immer so viel wie erforderlich, betonte er. Kinder müssten beispielsweise für eine MRT sediert werden, was sich gerade in den ersten Lebensjahren negativ auf die ZNS-Entwicklung auswirken könne. Daher müssten in der Pädiatrie immer auch die möglichen Langzeitfolgen von Diagnostik und Therapie bedacht werden, so Kager.
Psycho:Logisch: Lebensqualität nicht vernachlässigen
Der Psychologe Mag. András Acél, AKH Wien, erklärte, dass etwa ein Drittel der Patient:innen mit Weichteiltumoren psychologische Unterstützung benötigen.18 Zu den Faktoren, die die psychische Belastung beeinflussen, gehören laut Acél die Krankheit selbst (Tumorart/-lokalisation, Stadium/Prognose, Therapie), die Krankheitsverarbeitung (Kontrollverlust, Selbstbild/Integrität, Vorbelastung) und das soziale Netzwerk, in dem die Patient:innen leben (Einstellung, Religion etc.).
Im Krankheitsverlauf variiert die psychische Belastung. Besonders hoch sei sie zwischen der Diagnose und der Behandlung, vor einer Operation und bei einem Rezidiv, sagte der Psychologe. Die Belastung ende nicht nach erfolgreicher Therapie, da auch in der Nachsorge oft die Rezidivangst bestehen bleibe, so Acél. In der Palliativsituation stünden dagegen die körperlichen Beschwerden im Vordergrund.
In jeder Phase stehen unterschiedliche Interventionen zur Verfügung. In der Akutphase nach der Diagnose und bei einem Rezidiv geht es primär um eine Krisenintervention, um die Betroffenen zu stabilisieren und zu entlasten. Während der Therapie werden Screeningverfahren eingesetzt, um den Bedarf der Patient:innen zu klären. Ziele sind, die Selbstwirksamkeit zu fördern, die Krankheitsverarbeitung zu unterstützen und die Belastung zu reduzieren, zum Beispiel durch Entspannungstechniken. In der Palliativphase geht es vor allem um die Beziehungsarbeit, kombiniert beispielsweise mit Entlastungsgesprächen. Dazu zählt auch die Betreuung der Angehörigen.
Quelle:
Cancer Update CCC Vienna: Muskuloskelettale Tumoren – Sarkome, 9. April 2024, Wien und online
Literatur:
1 WHO Classification of Tumours of Soft Tissue and Bone, 5th ed. 2020 2 Noebauer-Huhmann IM et al.: Eur Radiol 2023; doi: 10.1007/s00330-023-10425-5 (online ahead of print) 3 Segen’s Medical Dictionary. © 2012 Farlex, Inc. 4 Noria S et al.: J Bone Joint Surg Am 1996; 78(5): 650-5 5 Enneking WF et al.: Clin Orthop Relat Res 1980; 153: 106-20 6 Dickinson IC et al.: ANZ J Surg 2006; 76(3): 104-9 7Kainhofer V et al.: Eur J Surg Oncol 2016; 42(6): 899-906 8 Ahmad R et al.: Oncologist 2016; 21(10): 1269-76 9 Harati K et al.: Oncologist 2017; 22(11): 1400-10 10 Harati K, Lehnhardt M: Innov Surg Sci 2017; 2(4): 165-70 11 Lazarides AL et al.: Clin Orthop Relat Res 2019; 477(4): 718-27 12 Wente MN et al.: Langenbecks Arch Surg 2007; 392(1): 83-93 13 Potkrajcic V et al.: Radiol Oncol 2021; 55(4): 459-66 14 Pasquali S et al.: J Clin Oncol 2018; 36(15_suppl): https://doi.org/10.1200/JCO.2018.36.15_suppl.11518 15 Pautier P et al.: Ann Oncol 2021; 32(suppl_5): S1283-346 16 Schöffski P et al.: J Clin Oncol 2015; 33(18_suppl): https://doi.org/10.1200/jco.2015.33.18_suppl.lba10502 17Oberoi S et al.: Pediatr Blood Cancer 2023; 70(Suppl 6): e30583 18 Eichler M et al.: Front Endocrinol (Lausanne) 2023; 14: 1166838
Das könnte Sie auch interessieren:

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

ASH 2020 – Highlights zu den aggressiven Lymphomen
Highlight-Themen der virtuellen ASH-Jahrestagung im Dezember 2020 waren an erster Stelle die Immunonkologika in all ihren Variationen, aber auch Beispiele für innovative Sequenztherapien ...

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...