
Update Ataxien
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Ataxien sind eine heterogene Gruppe seltener neurologischer Erkrankungen unterschiedlicher Ätiologie. Speziell die Gruppe der genetischen Ataxien hat sich in den letzten Jahren aufgrund der Fortschritte auf dem Gebiet der Genetik kontinuierlich vergrößert. Neue pathophysiologische Erkenntnisse sowie daraus resultierende experimentelle Therapieansätze lassen mit Zuversicht in die Zukunft blicken.
Keypoints
-
Die Ataxien werden in erworbene, sporadische und genetische Ataxien (Heredoataxien) unterteilt. Neben dem obligatorischen Leitsymptom Ataxie kommen häufig andere neurologische sowie gelegentlich auch nicht neurologische Manifestationen vor.
-
Die Klinik inklusive Zusatzuntersuchungen sowie eine ausführliche Familienanamnese bestimmen das weitere diagnostische Vorgehen, im Speziellen die sinnvolle genetische Untersuchung/Testung.
-
Aktuell stehen lediglich symptomatische und neurorehabilitative Therapien bei den Heredoataxien zur Verfügung.
-
In Zukunft dürften neue, innovative Therapieansätze mittels Genschere oder Antisense-Oligonukleotiden bzw. Modifikatoren pathophysiologischer Mechanismen den progredienten Krankheitsverlauf günstig beeinflussen.
Der Begriff Ataxie stammt aus dem Griechischen und bedeutet Unordnung bzw. Unregelmäßigkeit und bezeichnet in der Neurologie einerseits ein Symptom mit Störung der Bewegungskoordination, andererseits aber auch eine spezielle Gruppe von heterogenen, nicht fokalen Erkrankungen des Kleinhirns und dessen Verbindungen mit dem Leitsymptom Ataxie.
Einteilung
Anatomisch
Aus neurologischer Sicht wird je nach Lokalisation der betroffenen Körperregionen zwischen einer Extremitätenataxie (bestehend aus Dysmetrie, Intentionstremor und Dysdiadochokinese) und einer axialen Ataxie (mit Rumpf-, Stand- und Gangataxie) unterschieden. Diese Symptome können in Kombination oder auch (selten) isoliert auftreten. Aus neuroanatomischer Sicht wird zwischen zerebellären Ataxien mit prädominanter Affektion des Kleinhirns und spinalen oder sensiblen Ataxien mit prädominanter Affektion des Rückenmarks diskriminiert.
Ätiologisch
Gemäß ihrer Ätiologie werden Ataxien in erworbene, sporadisch-degenerative und hereditäre Ataxien eingeteilt.
Erworbene Ataxien
Erworbene Ataxien sind entweder auf exogene Noxen (mit Alkohol als häufigster Noxe, Intoxikation/Überdosierung von Phenytoin), Vitaminmangelzustände, Paraneoplasien (im Rahmen einer paraneoplastischen Kleinhirndegeneration) oder parainfektiöse Ursachen sowie Ataxien durch autoimmunologische Prozesse (z.B. bei GAD- und Gliadin-Autoantikörpern) zurückzuführen.
Sporadisch-degenerative Ataxien
Die Gruppe der sporadisch-degenerativen Ataxien umfasst einerseits die Multisystematrophie vom zerebellären Typ (MSA-C) und andererseits die idiopathische, spät beginnende zerebelläre Ataxie des Erwachsenenalters (ILOCA – „idiopathic late-onset cerebellar ataxia“, SAOA – „sporadic adult-onset ataxia“), welche im Endeffekt eine Ausschlussdiagnose darstellt.
Hereditäre Ataxien
Die Gruppe der hereditären Ataxien wird gemäß ihrem genetischen Hintergrund in autosomal-dominante, welche langläufig als spinozerebelläre Ataxien bezeichnet werden, autosomal-rezessive sowie die selteneren X-chromosomal vererbten Ataxien unterteilt. Zudem tritt das Symptom Ataxie auch bei einigen mitochondrial vererbten neurologischen Erkrankungen auf.
Klinische Manifestation
Genaue Prävalenzdaten für Ataxien im Allgemeinen fehlen, unter den erworbenen Ataxien macht jedoch die Alkohol-bedingte chronische zerebelläre Ataxie den Großteil der Fälle aus.
Autosomal-rezessive Ataxien
Die häufigste hereditäre Ataxie ist die autosomal-rezessiv vererbte Friedreich-Ataxie mit einer Prävalenz von 2–4/100000. Ursächlich ist eine „GAA triplet-repeat expansion“ im 1. Intron des Frataxin-Gens (FXN), was in weiterer Folge aufgrund beeinträchtigter Transkription zu verminderten Frataxin-Proteinspiegeln führt.1 In weiterer Folge wird eine mitochondriale Dysfunktion basierend auf einer Beeinträchtigung der mitochondrialen Atmungskette und damit erhöhtem oxidativem Stress postuliert.2 Klinisch-neurologisch zeigt sich eine spinale Ataxie mit progredienter Stand- und Gangataxie, Dysarthrie, Extremitätenataxie. Zu den extra-neuronalen Manifestationen zählen Skoliose, Pes equinovarus, Kardiomyopathie und Diabetes mellitus. Neuroanatomisch korreliert die Klinik entsprechend einer Degeneration der Spinalganglienzellen, der Hinterstränge sowie der spinozerebellären Verbindungen.
Weitere rezessiv vererbte Ataxien wie zum Beispiel die Ataxia teleangiectatica (AT, Louis-Bar-Syndrom) oder die Ataxien mit okulomotorischer Apraxie (AOA) (Typ I und II) kommen deutlich seltener vor und manifestieren sich bereits häufig in jungen Jahren (Kindheit, Adoleszenz). Pathophysiologisch finden sich beeinträchtigte DNA-Reparaturmechanismen, was vor allem im Falle der AT zu einer höheren Rate an Tumoren bei Betroffenen führt. Eine Ausnahme bezüglich des Manifestationsalters von rezessiven Ataxien stellt die erst in den letzten Jahren zunehmend erforschte SYNE1-Ataxie dar. Die SYNE1-Ataxie manifestiert sich durchwegs im Erwachsenenalter mit einem heterogenen klinischen Bild, das von einem rein zerebellären Phänotyp bis hin zu einer zusätzlichen Affektion der Pyramidenbahn und Hirnstammareale reicht.3
X-chromosomal-rezessiv vererbte Ataxien
Bei den X-chromosomal-rezessiv vererbten Ataxien ist das fragile X-assoziierte Tremor-Ataxie-Syndrom (FXTAS) zu nennen. Genetisch handelt es sich hierbei auch um eine Trinukelotid-Repeat-Erkrankung mit einer „CGG repeat expansion“ in der nicht codierenden 5‘-Region des FMR1-Gens von bis zu 200 Wiederholungen.4 Bei einer Repeat-Länge von über 200 CGG-Wiederholungen besteht ein fragiles X-Syndrom, welches die häufigste erbliche Ursache einer kognitiven Beeinträchtigung bei Knaben ist.5 Somit kann beim FXTAS von einer „Prämutation“ gesprochen werden, welche im höheren Lebensalter vor allem bei Männern (da X-chromosomal vererbt) zu einem Symptomenkomplex, bestehend aus Ataxie, Intentionstremor und kognitiver Dysfunktion, führt. Parkinsonismus und Neuropathie sind häufige fakultative Symptome. Frauen können je nach X-Chromosom-Inaktivierungsstatus (Lyon-Hypothese) auch an einem FXTAS erkranken. Häufiger findet sich bei Anlageträgerinnen eine polyzystische Ovarialinsuffizienz, während kognitive Defizite deutlich seltener als bei Männern auftreten. Pathophysiologisch führt die Repeat-Verlängerung zu einer Beeinträchtigung der Transkription. Je nach Repeat-Länge resultiert daraus entweder eine vermehrte Transkription von FMRP1-mRNA (inklusive abnormer mRNA) aufgrund von RAN („repeat-associated non ATG transcription“) oder wie im Falle des FXS (CGG>200) eine verminderte bzw. fehlende Transkription.
Spinozerebelläre Ataxien
Die spinozerebellären Ataxien (SCAs) umfassen eine äußerst heterogene und stetig wachsende Gruppe von autosomal-dominant vererbten Ataxien. Bei diesen Erkrankungen reicht bereits ein mutiertes Allel aus, um einen Phänotyp hervorzubringen. Somit besteht eine 50%-Wahrscheinlichkeit, das mutierte Allel und somit die Erkrankung weiterzugeben. Wie der Terminus „spinozerebellär“ impliziert, sind neben dem Kleinhirn (Cerebellum) auch das Rückenmark bzw. entsprechende Verbindungen zum Kleinhirn betroffen. Tatsächlich sind jedoch je nach SCA-Typ weitere zentralnervöse Strukturen wie z.B. die Basalganglien oder der Hirnstamm betroffen. Zudem findet man bei einigen SCAs auch lediglich ein rein isoliertes zerebelläres Syndrom.
Aktuell sind über 40 unterschiedliche SCAs bekannt und es kommen aufgrund des raschen Fortschritts in der Genetik kontinuierlich neue SCAs hinzu. Populationsbasierte Natural-History-Studien ergaben Prävalenzen für SCAs bis 5,6/100000.6
Bei den SCAs werden zwei Hauptgruppen unterschieden, nämlich die „repeat expansion SCAs“ und SCAs aufgrund von sogenannten konventionellen Mutationen (Punkt-, Missense-Mutationen etc.). Die erste Gruppe macht in Summe den Großteil aller SCAs aus und ist die am besten erforschte Gruppe. „Repeat expansion SCAs“ gemeinsam ist eine pathologische Expansion einer Nukleotidabfolge, meistens ein CAG-(Tri-)Nukleotid. Sofern die Repeat-Verlängerung in einem codierenden Abschnitt des entsprechenden Gens liegt, wird CAG in Glutamin translatiert und es führt zu einer verlängerten Abfolge von Polyglutaminen im entsprechenden Protein. Daher werden diese Erkrankungen auch Polyglutaminerkrankungen (Poly-Q, das Q ist der biochemische Code für die Aminosäure Glutamin) genannt.7 Zu den „klassischen“ CAG-Trinukleotid-Expansions-SCAs zählen SCA1, SCA2, SCA3/Machado-Joseph-Erkrankung, SCA6, SCA7 und SCA17 sowie die DRPLA. Trinukleotid-Expansionen in nicht codierenden Genregionen findet man bei der SCA8 (CTG-Expansion)8 sowie der SCA12 (CAG-Expansion)9. Es sind zumindest vier SCAs (SCA10, -31, -36 und -37) aufgrund von Repeat-Expansionen in intronischen (somit nicht proteincodierenden) Genregionen bekannt. Diesen ist gemein, dass die Repeat-Zahl deutlich über jener bei den „klassischen“ exonischen Repeat-Expansionen liegt und im Falle der SCA10 zum Beispiel bis 4500 Repeats umfassen kann.10
Weltweit sind die SCA3/Machado-Joseph-Erkrankung gefolgt von der SCA2 und der SCA6 die häufigsten SCAs. Die geografische Verteilung der unterschiedlichen SCAs lässt Rückschlüsse auf sogenannte „Founder“-Effekte, wie dies zum Beispiel bei der SCA2 in Kuba oder der SCA3/Machado-Joseph-Erkrankung in Portugal/Brasilien der Fall ist, zu. So findet man für die SCA2 eine Prävalenz von 40/100000 in Kuba,11 für die SCA3/Machado-Joseph-Erkrankung auf der Azoreninsel Flores eine Prävalenz von 1/239.12
Den „repeat expansion SCAs“ ist gemeinsam, dass die zugrunde liegende Mutation instabil ist und es sowohl zu einer Zunahme als auch zu einer Abnahme der Repeats kommen kann. Dies kann gewebs- oder zellspezifisch der Fall sein oder auch über Generationen hinweg passieren, wobei es hier im Regelfall zu einer Zunahme der Repeat-Anzahl bei den Nachkommen kommt, was als Antizipation bezeichnet wird. Diese Zunahme der Repeat-Anzahl, sei sie maternalen oder paternalen Ursprungs, resultiert sowohl in einem früheren Erkrankungsbeginn als auch einer schwerer ausgeprägten Krankheitsmanifestation.
Die andere große Gruppe der SCAs umfasst jene, welche aufgrund von konventionellen Mutationen (Punktmutationen, Deletionen, Insertionen) hervorgerufen werden. Im Gegensatz zu den „repeat expansion SCAs“ liegt deren Häufigkeit deutlich unter jener der klassischen „repeat expansion SCAs“.13 Zu den zwei häufigsten auf konventionellen Mutationen beruhenden SCAs zählen die SCA14 und SCA28, welche in europäischen Kohortenstudien ca. 1–4% aller SCAs ausmachen.14
Insgesamt ist jedoch zu berücksichtigen, dass bei bis zu 50% aller SCAs (also genetisch suszipierten Erkrankungen, welche einem dominanten Erbgang folgen und klinisch ein spinozerebelläres Syndrom aufweisen) aktuell kein genetischer zugrunde liegender Defekt gefunden werden kann.
Einblicke in mögliche zugrunde liegende pathophysiologische Mechanismen gelangen überwiegend durch entsprechende tier- beziehungsweise zellbasierte Modelle für „repeat expansion SCAs“. Aufgrund der genetischen Grundlage eines autosomal- dominanten Erbganges reicht bereits ein mutiertes Allel aus, um einen Phänotyp zu verursachen. Dies kann durch eine Toxizität des mutierten Proteins als auch durch eine resultierende Haploinsuffizienz (dominant-negativ) geschehen. Der genaue Pathomechanismus für die einzelnen Entitäten ist nach wie vor unbekannt, jedoch scheinen einige gemeinsame pathophysiologische Mechanismen als mögliche Ziele für zukünftige Therapien zu bestehen. Diese reichen von einer veränderten Proteinstruktur, welche zu neuronalen (meist nukleären) Aggregationen bzw. Einschlüssen (Polyglutamin-SCAs) neigen, über eine durch das mutierte Protein veränderte Interaktion mit weiteren Proteinen bis zu bioenergetischen Defiziten auf mitochondrialer Ebene (z.B. SCA28)15 und Ionenkanaldysfunktionen (z.B. SCA6)16. Speziell bei nicht codierenden „repeat expansion SCAs“ konnte zudem eine RNA-vermittelte Toxizität experimentell nachgewiesen werden.
Natural-History-Studien bei spinozerebellären Ataxien wie die europäische multizentrische RISCA-Studie („prospective study of individuals at risk for SCA1 SCA2, SCA3, SCA6, SCA7“) haben gezeigt, dass bereits vor Auftreten einer progredienten Ataxie andere, teils subtile neurologische und/oder radiologische Befunde zu erheben sind. Neben der allen SCAs gemeinsamen progredienten Ataxie als Hauptmerkmal bestehen je nach SCA-Subtyp unterschiedliche neurologische Zusatzsymptome, wobei die Überlappung dieser zwischen den unterschiedlichen SCAs groß ist. Man findet Störungen der Optomotorik, Polyneuropathien, Myoklonien, parkinsonistische und choreoathetotische Symptome bis hin zu retinaler Degeneration (SCA7) und kognitiven Defiziten.
Genetische Diagnostik
Bei Vorliegen eines progredienten ataktischen Syndroms und nach Ausschluss erworbener Ursachen, wie oben bereits besprochen, sowie einer positiven Familienanamnese wird eine zügige genetische Testung empfohlen (Abb. 1). In einer ersten Runde soll im Falle einer positiven Familienanamnese vereinbar mit autosomal-dominantem Erbgang auf klassische Repeat-Expansionen (Polyglutamin-)SCAs unter Berücksichtigung regionaler Gegebenheiten (Stammbaum, Herkunft) getestet werden, da diese Tests relativ einfach, kostengünstig und weit verbreitet angeboten werden. Im Falle einer negativen ersten Testrunde kann in einer zweiten Testrunde auf mögliche nicht codierende „repeat expansion SCAs“ getestet werden beziehungsweise im weiteren Verlauf ein „whole-exom sequencing“ durchgeführt werden. Hierbei ist jedoch zu beachten, dass alle Repeat-Expansionen im Rahmen des „whole-exome sequencing“ bislang nicht detektiert werden. Im Falle eines suspizierten rezessiven Erbganges und entsprechender Klinik mit vornehmlich spinaler Ataxie sollte an erster Stelle die genetische Testung auf Friedreich-Ataxie (FRDA) erfolgen, da diese die häufigste Heredoataxie überhaupt ist. Je nach zusätzlichen Symptomen kann die genetische Diagnostik bezüglich AOA1/2, AT oder SYNE1-Mutationen erweitert werden. Verschiedene SCA-/Ataxie-Panels werden kommerziell angeboten und ständig erweitert. Auch bei sporadischen, also ohne positive beziehungsweise konklusive Familienanamnese auftretenden progredienten Ataxien ist durchaus in der diagnostischen Abklärung eine genetische Testung für zumindest die klassischen Polyglutamin-SCAs sowie auf FRDA, FXTAS und SYNE1 anzudenken. Bei bis zu einem Viertel der Patienten mit sporadischen Ataxien konnte in europäischen und amerikanischen Studien eine genetische Ursache gefunden werden, wobei die SCA6 am häufigsten in dieser Gruppe vertreten war.
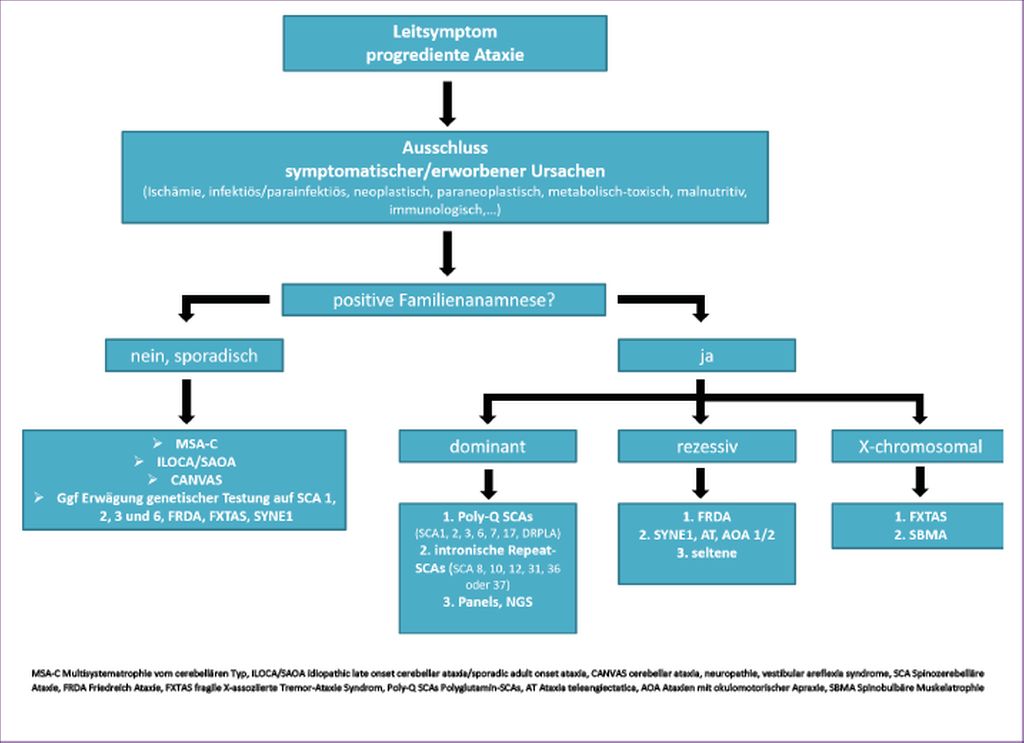
Abb. 1: Abklärung der Ursachen bei Vorliegen eines progredienten ataktischen Syndroms
Erst rezent wurde bei einem bereits in den 90er-Jahren erstmals beschriebenen Symptomkomplex bestehend aus spät beginnender zerebellärer Ataxie, Neuropathie und Vestibulopathie, bekannt als CANVAS („cerebellar ataxia, neuropathy and vestibular areflexia syndrome“), bei einem Teil der Erkrankten (der familiären Form) eine ursächliche Mutation im Sinne einer biallelischen (somit rezessiven) intronischen Pentanukleotid-Expansion im RFC1(„replication factor C subunit 1“)-Gen entschlüsselt.17 In einer Population von Patienten mit einer spät beginnenden sporadischen zerebellären Ataxie wurde eine Häufigkeit jener RFC1-Mutation, die deutlich höher im Falle einer Kombination von zerebellärer Ataxie, Neuropathie und Vestibulopathie war, von 22% gefunden.18 Dieses Beispiel demonstriert die Fortschritte auf dem Gebiet der Genetik bei Ataxien, welche auch klinische Auswirkungen haben. So sollte man bei spät beginnenden zerebellären Ataxien eine RFC1-Mutation differenzialdiagnostisch in Erwägung ziehen – eine kommerzielle, genetische Testung ist jedoch bislang nicht etabliert.
Fehlende Therapieoptionen
In deutlichem Gegensatz zu den Fortschritten, was die genetische Entschlüsselung und das pathophysiologische Verständnis der Heredoataxien betrifft, stehen fehlende Fortschritte bei Therapieoptionen. Dies mag einerseits der Heterogenität dieser Erkrankungsgruppe zugrunde liegen, zum anderen jedoch auch der niedrigen Prävalenz. Wie bei anderen neurodegenerativen Erkrankungen fehlen valide Biomarker, welche mögliche Therapieeffekte entsprechend abbilden, vor allem in der frühen „präataktischen“ Erkrankungsphase. Aktuelle Ataxieskalen, von denen vor allem die SARA („scale for the assessment and rating of ataxia“)19 eine häufig und einfach anzuwendende Skala darstellt, zeigen vor allem in späten Krankheitsstadien einen sogenannten „ceiling effect“. Dies bedeutet, dass in fortgeschrittenen Krankheitsstadien ein möglicher Therapieeffekt im Sinne einer verminderten oder gar fehlenden Krankheitsprogression nicht adäquat aufgrund unterschiedlicher Gewichtung der betroffenen Systeme (Ataxie der unteren Extremität – Gang versus Ataxie der oberen Extremität) in der entsprechenden Rating-Skala abgebildet wird. Nichtsdestotrotz wurden in den letzten Jahren zumindest einige Pilotstudien an einer kleinen Anzahl von Ataxiepatienten durchgeführt. Unter anderem zeigte der NMDA-Rezeptor-Antagonist Riluzol, welcher als Therapie für die amyotrophe Lateralsklerose zugelassen ist, in einer heterogenen Gruppe von Heredoataxien einen Benefit im Sinne einer Verbesserung des SARA-Wertes verglichen mit Placebo.20, 21 Limitierend an der durchgeführten Studie war die kleine und heterogene Studienpopulation. Weitere Kandidatensubstanzen, unter anderem das Anti-Konvulsivum Valproinsäure oder Lithiumcarbonat, konnten keine überzeugende Evidenz im Hinblick auf eine positive Krankheitsmodifizierung/-beeinflussung liefern. Somit stellen symptomatische Therapien vor allem der Nicht-Ataxiesymptome (Neuropathie, Parkinsonsymptome, Chorea, kognitive Beeinträchtigung, Depression etc.) eine wesentliche Säule der Therapie dar. Ergänzend tragen physio-, ergo- und logopädische Therapien wesentlich zur Aufrechterhaltung der Selbstständigkeit und Mobilität und folglich zur Lebensqualität bei.
Kandidatensubstanztestung vornehmlich an humanen neuronalen Zellkulturen, welche aus Patienten-induzierten pluripotenten Stammzellen (iPSC) generiert werden, ermöglicht eine rasche und großflächige Testung verschiedenster Substanzen. Epigenetische Modifikatoren, speziell bei den nicht codierenden „repeat expansion SCAs“, dem FXS oder der Friedreich-Ataxie, können auf Transkriptionsebene die Genexpression regulieren. Vor allem lassen jedoch rezente Forschungen auf dem Gebiet der molekularen Therapie, sei es mittels CRISPR/Cas9 zum Entfernen bzw. zur Korrektur des mutierten Allels, RNA-Interference mittels siRNA („small interfering RNAs“) oder ASOs („antisense-oligonucleotides“) zur Verhinderung des dominant-negativen/toxischen Effekts des mutierten Gens, zuversichtlich und hoffnungsvoll in die Zukunft der Therapien der Heredoataxien blicken.
Definitionen
maternal – paternal: mütterlicherseits – väterlicherseits
Gen: Abschnitt auf der DNA, welcher die Grundinformationen zur Herstellung von biologisch aktiver mRNA enthält und folglich für die Eigenschaften eines Individuums verantwortlich ist
Allel: Merkmalsausprägung eines Gens. Jedes Gen kommt in 2 Allelen-Varianten/Merkmalsausprägungen vor (ein väterliches und ein mütterliches).
Genotyp: Gesamtheit aller Gene eines Organismus
Phänotyp: morphologische und physiologische Merkmalsausprägungen aufgrund eines entsprechenden Genotyps
Transkription: Umschreiben eines Gens von DNA (Desoxyribonukleinsäure) in mRNA (Messenger-Ribonukleinsäure)
Translation: Synthese von Proteinen aus der mRNA
Exon: Teil eines Gens, das nach dem Spleißen (Prozessierung der mRNA) übrig bleibt und in weiterer Folge translatiert wird
Intron: Teil eines Gens, das nach dem Spleißen abgebaut wird und somit in der Folge nicht translatiert wird
Triplet-repeat: Wiederholung von Trinukleotiden in der DNA.
RAN: „repeat-associated non ATG transcription“, nicht klassische und somit alternative Transkription von Genabschnitten mit Nukleotidexpansionen
Sanger-Sequenzierung: klassische und Goldstandard-Methode der Sequenzierung (=Bestimmung der Abfolge der Nukleotide) von DNA/Genabschnitten. Geeignet für Einzelgenanalysen.
Whole-genome sequencing (WGS): Methode des „next-generation sequencing“ (NGS), einer relativ neuen Methode der massiven parallelen Sequenzierung von DNA-Abschnitten, mit der aufgrund des hohen Durchsatzes das gesamte Genom in wenigen Tagen sequenziert werden kann
Whole-exome sequencing (WES): relativ neue Methode des „next-generation sequencing“, wobei nur die Exons (codierenden Genabschnitte) vervielfältigt und sequenziert werden
Haploinsuffizienz: Der Menschen hat, wie andere diploide Organismen, auf seinen homologen Chromosomen zwei Kopien (Allele) eines Gens. Im Falle eines mutierten Allels (wie z.B. bei autosomal-dominant vererbten Erkrankungen), reicht das nicht mutierte (=Wildtyp) Gen in seinem haploiden Zustand nicht aus, um einen regelrechten Phänotyp zu bewirken.
Founder-Effekt: Gründereffekt; beschreibt das Phänomen, dass in einer bestimmten, isolierten (Gründer-)Population eine geringe Anzahl von Allelen (Merkmalsausprägungen) vorkommt und somit im Falle eines mutierten Allels autosomal-dominant vererbte Erkrankungen in jener Population überdurchschnittlich häufig auftreten.
RNA-Interferenz: zielgerichteter Mechanismus, welcher aufgrund einer Wechselwirkung von kurzen komplementären RNA-Stücken mit einer spezifischen mRNA diese stilllegt und somit die Expression eines Gens abschalten kann.
ASO: Antisense-Oligonukleotide sind kurzkettige synthetisch hergestellte einzelsträngige Nukleinsäureketten, deren Basensequenz komplementär (also entgegengesetzt) zu einer funktionalen mRNA ist, was in der Folge die Translation verhindert.
Literatur:
1 Campuzano V et al.: Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996; 271: 1423-7 2 Pandolfo M: Friedreich ataxia: the clinical picture. J Neurol 2009; 256(Suppl 1): 3-8 3 Synofzik M et al.: SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain 2016; 139(Pt 5): 1378-93 4 Hagerman R, Hagerman P: Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. The Lancet Neurology 2013; 12(8): 786-98 5 Hagerman RJ et al.: Fragile X syndrome. Nat Rev Dis Primers 2017; 3: 17065 6 Klockgether T et al.: Spinocerebellar ataxia. Nat Rev Dis Primers 2019; 5(1): 24 7 Paulson HL et al.: Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat Rev Neurosci 2017; 18(10): 613-26 8 Mosemiller AK et al.: Molecular genetics of spinocerebellar ataxia type 8 (SCA8). Cytogenet Genome Res 2003; 100(1-4): 175-83 9 Holmes SE et al.: Expansion of a novel CAG trinucleotide repeat in the 5‘ region of PPP2R2B is associated with SCA12. Nat Genet 1999; 23(4): 391-2 10 Matsuura T et al.: Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 2000; 26(2): 191-4 11 Orozco Diaz G et al.: Autosomal dominant cerebellar ataxia: clinical analysis of 263 patients from a homogeneous population in Holguin, Cuba. Neurology 1990; 40(9): 1369-75 12 Bettencourt C et al.: Analysis of segregation patterns in Machado-Joseph disease pedigrees. J Hum Genet 2008; 53(10): 920-923 13 Durr A: Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010; 9(9): 885-94 14 Hersheson J et al.: The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum Mutat 2012; 33(9): 1324-32 15 Cagnoli C et al.: SCA28, a novel form of autosomal dominant cerebellar ataxia on chromosome 18p11.22-q11.2. Brain 2006; 129(Pt 1): 235-42 16 Riess O et al.: SCA6 is caused by moderate CAG expansion in the alpha1A-voltage-dependent calcium channel gene. Hum Mol Genet 1997; 6(8): 1289-93 17 Cortese A et al.: Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 2019; 51(4): 649-58 18 Aboud Syriani D et al.: Prevalence of RFC1-mediated spinocerebellar ataxia in a North American ataxia cohort. Neurol Genet 2020; 6(3): e440 19 Schmitz-Hubsch T et al.: Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006; 66(11): 1717-20 20 Ristori G et al.: Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology 2010; 74(10): 839-45 21 Romano S et al.: Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2015; 14(10): 985-91
Das könnte Sie auch interessieren:

Wichtige Laborparameter bei Epilepsie: ein aktueller Überblick
Laborkontrollen sind bei Epilepsien aus verschiedensten Gründen erforderlich. Anfallssupprimierende Medikamente können Störwirkungen auf unterschiedliche Organsysteme haben, die unter ...

Alzheimer: laufende klinische Studien, State of the Art der Biomarkerdiagnostik und gemischte Pathologien
Die Forschung an neuen Therapien für die Alzheimerkrankheit (AD) erfährt ein nie dagewesenes Momentum. Auf der internationalen Alzheimer- und Parkinsonkonferenz AD/PD in Wien gab es ...

Nahrungsergänzungsmittel bei ME/CFS: neue Hoffnung oder falsche Versprechen?
Chronische Erschöpfung, die nicht vergeht, Schmerzen, Konzentrationsprobleme, ein Leben in Zeitlupe. Myalgische Enzephalomyelitis/Chronisches Fatigue-Syndrom (ME/CFS) betrifft weltweit ...