
Neue Diagnosekriterien und Therapiestrategien
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die amyotrophe Lateralsklerose (ALS) ist eine progrediente neurodegenerative Erkrankung, die mit einer Überlebenszeit von 2–4 Jahren noch immer tödlich verläuft. Es sind primär das 1. und 2. Motoneuron (MN) in einem variablen Ausmaß betroffen, wobei abseits des motorischen Systems häufig auch extramotorische neuronale Zentren involviert sind. Wesentliche zugrunde liegende Pathomechanismen sind nach wie vor ungeklärt. Eine Reihe neuer Erkenntnisse hat jedoch in den vergangenen Jahren zur Entwicklung innovativer Erkrankungsmodelle und zur Definition neuer Diagnosekriterien geführt. In Summe haben diese Tools das Potenzial, die Therapieentwicklung voranzutreiben und die klinische Versorgung zu optimieren.
Diagnosekriterien für die klinische Routine

Tab. 1: Gold-Coast-Kriterien
Für die Diagnose der ALS sind in erster Linie der klinische Nachweis einer Motoneuronschädigung sowie ein progredienter Erkrankungsverlauf essenziell. Wichtig ist auch der Ausschluss möglicher Differenzialdiagnosen, der je nach klinischer Befundkonstellation verschiedene Untersuchungsmodalitäten einschließt. Die bisherigen El-Escorial- bzw. Awaji-Diagnosekriterien wurden ursprünglich für Studienzwecke entwickelt und differenzierten unter Berücksichtigung klinischer und elektromyografischer Parameter die Diagnosewahrscheinlichkeiten einer „möglichen“, „wahrscheinlichen“ und „definitiven“ ALS. Für die klinische Praxis waren diese Kriterien ungeeignet, da sie einerseits aufgrund der restriktiven Gestaltung oft erst in späteren Erkrankungsstadien eine definitive Diagnose erlaubten und andererseits atypische Formen nicht erfassten. Die neuen Gold-Coast-Diagnosekriterien (Tab. 1) dagegen erlauben eine sichere und frühzeitigere Diagnose, insbesondere bei atypischen Formen wie der bulbären ALS oder Sonderformen wie der progressiven Muskelatrophie.1,2 Zum ersten Mal liegen somit Diagnosekriterien vor, die sich sowohl für den Einsatz in der klinischen Routine als auch für die Rekrutierung von Patient*innen für klinische Studien eignen.
Das Erkennen von Paresemustern
Die ALS ist gekennzeichnet durch einen heterogenen Beginn der Erkrankung mit variabler Beteilung des 1. und 2. MN sowie mit Unterschieden in der Lokalisation der initialen Symptomatik, wonach eine spinale (ca. 80% der Fälle), eine bulbäre (ca. 15% der Fälle) sowie eine axiale Form (<5% der Fälle) differenziert werden. Trotz dieser Heterogenität gibt es aber auch klinische Charakteristika, die bei der häufigen spinalen Form Paresemuster erkennen lassen, deren Beachtung für die Diagnose wichtig ist.
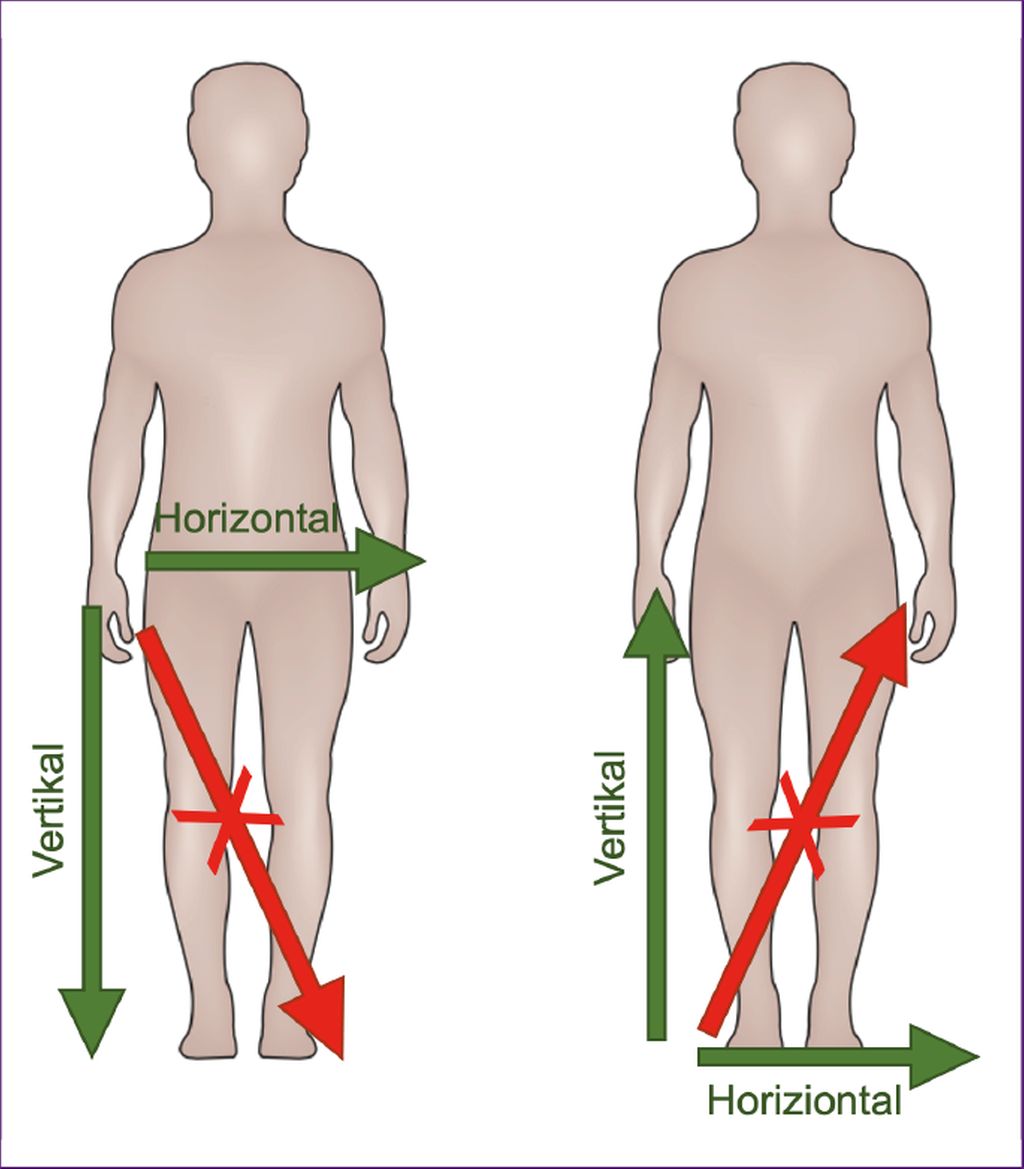
Abb. 1: Ausbreitungsmuster der Paresen
Fast immer beginnt die Parese fokal in einer distalen Extremität und breitet sich nach proximal und „horizontal“ auf die kontralaterale Seite aus (Abb. 1). Alternativ kann sich die Parese auch „vertikal“ von der Hand auf den ipsilateralen Fuß (bzw. vom Fuß auf die ipsilaterale Hand ausbreiten). Eine „diagonale“ Ausbreitung von der Hand auf den kontralateralen Fuß (bzw. vom Fuß auf die kontralaterale Hand) ist dagegen unwahrscheinlich und sollte eine gründliche differenzialdiagnostische Abklärung zur Folge haben. Abgesehen von den typischen Ausbreitungsmustern gibt es auch Prädilektionsmuster für die Paresen. An der Hand kommt es dabei typischerweise zu einer „Split hand“-Verteilung der Atrophie bzw. der Paresen, bei der die Thenarmuskulatur stärker betroffen ist als der Hypothenar. Weiters ist am Hand- und Fußgelenk die Extension früher und stärker betroffen als die Flexion, und im Ellbogen- und Kniegelenk ist die Flexion früher und stärker betroffen als die Extension. Die Prädilektion dieser Muskelgruppen bei der ALS wurde mit der stärkeren Ausprägung direkter kortikospinaler Efferenzen zu den spinalen Motoneuronen in den entsprechenden Segmenten in Verbindung gebracht.3
Das Problem des 1. Motoneurons

Tab. 2: Zeichen des 1. MN sind definiert als einer der folgenden Befunde
Die klinische Detektion von Schädigungszeichen des 1. Motoneurons in Form gesteigerter Reflexe oder einer Spastizität setzt ausreichend intakte spinale Motoneurone voraus4 und stellt folglich bei zusätzlicher Affektion des 2. MN eine besondere Herausforderung dar. Dies zeigt sich eindrucksvoll bei Patient*innen mit progressiver Muskelatrophie, bei denen klinisch ausschließlich Schädigungszeichen des 2. MN vorliegen. In neuropathologischen Untersuchungen konnte hier in 50% der Fälle sehr wohl auch eine Degeneration der kortikospinalen Efferenzen nachgewiesen werden.5 In den Gold-Coast-Diagnosekriterien sind die klinischen Parameter angeführt, die bei der klinischen Untersuchung zur Detektion von Schädigungszeichen des 1. MN herangezogen werden sollten (Tab. 2). Apparative Untersuchungen wie das MRT, die motorisch evozierten Potenziale und die transkranielle Magnetstimulation wurden dabei nicht in die Diagnosekriterien integriert, können aber sehr wohl supportiv herangezogen werden.
Neurofilamente als diagnostische und prognostische Biomarker
Neurofilamente (Nf) sind intrazelluläre Strukturproteine, die ausschließlich in Nervenzellen vorkommen und bei Zelluntergang im Serum und Liquor nachgewiesen werden können. Sie bestehen aus drei Untereinheiten, wobei bisher nur die leichten (NfL) und schweren (NfH) Neurofilament-Ketten als Biomarker genauer charakterisiert wurden. Bei der ALS werden besonders hohe Nf-Konzentrationen erreicht, sodass bei entsprechend hohen Grenzwerten eine Sensitivität und Spezifität zwischen 80 und 90% erzielt wird.6, 7 In der klinischen Praxis sind die Nf daher insbesondere bei der Abgrenzung zu Differenzialdiagnosen hilfreich. Aber auch bei der prognostischen Beurteilung spielen die Nf eine wichtige Rolle und übertreffen dabei auch herkömmliche Parameter wie die ALS-Funktionsskala (ALS Functional Rating Scale, ALSFRS-R8, 9), einen Score zur Beurteilung des Funktionsverlustes bei der ALS. Die Nf-Konzentration korreliert dabei direkt mit der Progressionsrate und indirekt mit dem Überleben.
Monogenetische ALS-Formen
Monogenetische Ursachen werden bei bis zu 60% der Patient*innen mit familiärer ALS10,11 und bei 5–10% der Patient*innen mit sporadischer ALS gefunden.12 Zu den häufigsten mit ALS ursächlich assoziierten Genen zählen dabei C9orf72, SOD1, FUS und TARDBP. Eine Empfehlung zur genetischen Testung wird aktuell nur für Patient*innen mit einer positiven Familienanamnese für ALS, Demenzen und psychiatrischen Erkrankungen empfohlen. Es muss jedoch auch bedacht werden, dass einerseits die Familienanamnesen nicht immer korrekt sind und andererseits auch eine unvollständige Penetranz bzw. eine De-novo-Mutation vorliegen kann. Daher muss im Aufklärungsgespräch auf den individuellen Standpunkt der Patient*innen eingegangen werden, insbesondere auch in Anbetracht laufender und anstehender Gentherapiestudien für SOD1, C9orf72- und FUS-Mutationen.
Neue Konzepte für klinische Studien
Trotz intensiver internationaler Forschungsbemühungen steht mit Riluzol nach wie vor das einzige von der Europäischen Arzneimittelagentur (EMA) für die ALS zugelassene Medikament zur Verfügung. Die wissenschaftlichen Erkenntnisse der vergangenen Jahre haben aber zu einer Optimierung der Therapiestrategien und zur Entwicklung effizienterer Studiendesigns geführt, die sich in Zukunft günstig auf die Medikamentenentwicklung auswirken wird (Tab. 3).
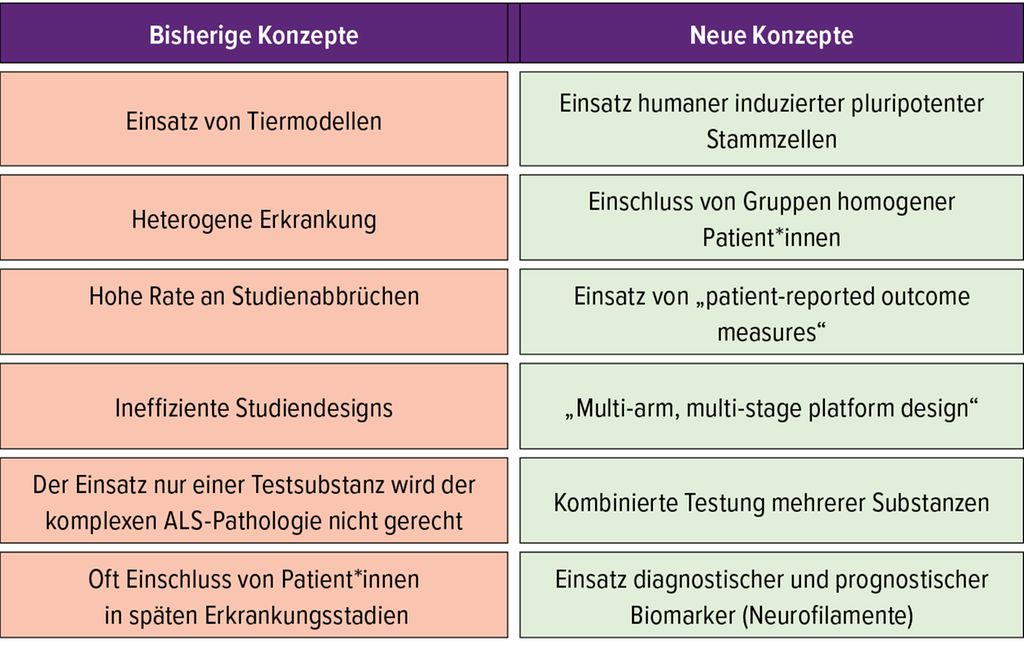
Tab. 3: Neue Ansätze zur Optimierung von Studienkonzepten
Induzierte pluripotente Stammzellen
Mit den induzierten pluripotenten Stammzellen (iPSC) steht ein neues Erkrankungsmodell zur Verfügung, das in der Grundlagenforschung den Weg für neue Therapien geebnet hat und das in präklinischen Studien gegenüber Tiermodellen den Vorteil hat, dass es sich dabei um ein humanes Modell handelt. Folglich scheinen auf iPSC basierende Resultate für die weitere Translation in den klinischen Bereich aussagekräftiger zu sein.13, 14
Einschluss homogener ALS-Verlaufsformen in klinische Studien
Die klinische und biologische Heterogenität der ALS wirkt sich negativ auf die Reproduzierbarkeit von Experimenten und Studien aus. Insbesondere der heterogene Erkrankungsverlauf bei ALS dürfte sich in klinischen Studien limitierend auf die tatsächliche Effektgröße von Testsubstanzen auswirken.15, 16 Es können hier inzwischen unterschiedliche Cluster von Progressionsraten ermittelt werden, deren Berücksichtigung in klinischen Studien zum Einschluss homogener Patient*innengruppen mit konsekutiver Steigerung der statistischen Power beitragen kann.
Patient-reported outcome measures
Für ALS-Patient*innen mit rasch progredienten funktionellen Einschränkungen stellen die regelmäßigen Besuche in den Studienzentren ein relevantes Problem dar. Die damit einhergehenden Belastungen können die Patient*innen zusätzlich ermüden und so die Kontrolluntersuchungen beeinflussen.16 Es wird geschätzt, dass >25% der Studienabbrüche auf die mit den Transporten assoziierten Belastungen zurückzuführen sind.17 „Patient-reported outcome measures“ können hier zu einer Entspannung der belastenden Situation für die Patient*innen führen und die Rate der Studienabbrüche reduzieren.
Multi-arm multi-stage design
„Multi-arm multi-stage“(MAMS)-Studien ermöglichen die gleichzeitige Untersuchung mehrerer Behandlungen gegenüber einer einzigen Kontrolle.18, 19 Dabei kann es sich entweder um unterschiedliche Medikamente, eine Kombination von Medikamenten oder um unterschiedliche Dosierungen desselben Medikaments handeln. MAMS-Studien haben ein adaptives Design, bei dem Studienarme frühzeitig abgebrochen werden und neue Studienarme dazukommen können. Dadurch ergibt sich eine Lenkung von Studienpatient*innen hin zu Studienarmen mit den wirksameren Medikamenten, wodurch wertvolle Zeit und Ressourcen gespart werden.
Fazit
Die intensive Forschungstätigkeit im Bereich der Motoneuronerkrankungen hat zu großen Fortschritten im Verständnis biologischer Zusammenhänge geführt. Die Translation dieser Erkenntnisse in den klinischen Bereich trägt zur Optimierung der Studienkonzepte bei und hat gleichzeitig das Potenzial, die Medikamentenentwicklung zu beschleunigen und die Patient*innenversorgung zu verbessern.
Literatur:
1 Hannaford A et al.: Diagnostic utility of gold coast criteria in amyotrophic lateral sclerosis. Ann Neurol 2021; 89: 979-86 2 Pugdahl K et al.: Gold coast diagnostic criteria increase sensitivity in amyotrophic lateral sclerosis. Clin Neurophysiol 2021; 132: 3183-9 3 Ludolph AC et al.: Pattern of paresis in ALS is consistent with the physiology of the corticomotoneuronal projections to different muscle groups. J Neurol Neurosurg Psychiatry 2020; 91: 991-8 4 Swash M: Why are upper motor neuron signs difficult to elicit in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry 2012; 83: 659-62 5 Ince PG et al.: Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology 2003; 60: 1252-8 6 Steinacker P et al.: Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry 2016; 87: 12-20 7 Verde F et al.: Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2019; 90: 157-64 8 Benatar M et al.: Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology 2020; 95: e59-e69 9 Thompson AG et al.: Multicentre appraisal of amyotrophic lateral sclerosis biofluid biomarkers shows primacy of blood neurofilament light chain. Brain Commun 2022; 4: fcac029 10 Muller K et al.: Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry 2018; 89: 817-27 11 Roggenbuck J et al.: Amyotrophic lateral sclerosis genetic access program: paving the way for genetic characterization of ALS in the clinic. Neurol Genet 2021; 7: e615 12 Renton AE et al.: State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014; 17: 17-23 13 Baxi EG et al.: Answer ALS, a large-scale resource for sporadic and familial ALS combining clinical and multi-omics data from induced pluripotent cell lines. Nat Neurosci 2022; 25: 226-37 14 Fujimori K et al.: Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat Med 2018; 24: 1579-89 15 Goyal NA et al.: Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve 2020; 62: 156-66 16 Kiernan MC et al.: Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat Rev Neurol 2021; 17: 104-18 17 Atassi N et al.: Analysis of start-up, retention, and adherence in ALS clinical trials. Neurology 2013; 81: 1350-5 18 Millen GC, Yap C: Adaptive trial designs: what are multiarm, multistage trials? Arch Dis Child Educ Pract Ed 2020; 105: 376-8 19 Wong C et al.: Motor neuron disease systematic multi-arm adaptive randomised trial (MND-SMART): a multi-arm, multi-stage, adaptive, platform, phase III randomised, double-blind, placebo-controlled trial of repurposed drugs in motor neuron disease. BMJ Open 2022; 12: e064173
Das könnte Sie auch interessieren:

Pharmakologische und neuromodulatorische Behandlungen des Clusterkopfschmerzes
Die Behandlung des chronischen Clusterkopfschmerzes steht noch heute vor großen Herausforderungen. Am 19. European Headache Congress (EHC) 2025 präsentierte Dr. Anja Petersen, ...

Ist die ketogene Diät eine Präzisionsmedizin?
Die ketogenen Ernährungstherapien sind etablierte Behandlungsformen bei Epilepsie. Während sie primär bei therapierefraktären pädiatrischen Epilepsien eingesetzt werden, finden sie ...

Neues aus der Alzheimer’s Disease Drug Development Pipeline
Mit der weltweiten Zulassung der Amyloidantikörper Lecanemab und Donanemab ist erstmals eine kausale Behandlung der Alzheimerkrankheit möglich geworden. Die Behandlung setzt an der ...