Phäochromozytom, Paragangliom und adrenokortikales Karzinom
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Phäochromozytome (Phäo) und Paragangliome (PGL) sind seltene neuroendokrine Tumoren. Die komplette chirurgische Entfernung ist die Therapie der Wahl. Aufgrund der potenziellen Malignität ist die Abklärung prä-, peri- und postoperativ von entscheidender Bedeutung und benötigt eine spezielle Infrastruktur und Wissen. Die Malignität kann nur durch den Beweis von Metastasen verifiziert werden. Daher sollten diese Tumorentitäten nur an speziellen Zentren wie an der Medizinischen Universität Wien behandelt werden.
Keypoints
-
Die Diagnostik von Nebennierentumoren besteht primär aus der Bestimmung von Katecholaminmetaboliten, Hormonen und der radiologischen/nuklearmedizinischen apparativen Diagnostik.
-
Eine konventionelle Bildgebung wie die CT ist obligat, aber oft nicht spezifisch genug und bedarf weiterer Diagnostik. Speziell die DOPA-PET/CT bei Phäo/PGL ist hier wichtig und sollte durchgeführt werden. Aber auch die FDG-PET/CT ist speziell für die Suche nach Metastasen bei allen Nebennierentumoren wichtig und besser als ein MIBG-Scan.
-
Die chirurgische Komplettentfernung ist die einzige kurative Therapie im lokalisierten Stadium.
-
Die Diagnostik und die Therapie von Nebennierenraumforderungen sollten in spezialisierten Zentren mit entsprechender Expertise erfolgen.
Phäo/PGL können sporadisch und im Rahmen von Keimbahnmutationen und hereditären Syndromen vorkommen. Daher wird empfohlen, alle PGL genetisch zu testen. Die Bestimmung von Plasma und Harnmetanephrinen sowie eine Bildgebung sollten jährlich durchgeführt werden. Die Nachsorge erfolgt mindestens 10 Jahre.
Im Vergleich zum Phäo, als neuroendokriner Tumor des Nebennierenmarks, entspringen adrenokortikale Karzinome (ACC) der Nebennierenrinde. Es handelt sich um extrem seltene und aggressive Tumoren, die einer vollumfassenden Abklärung aus Bildgebung und Hormonstatus bedürfen. Lokal begrenzt ist die vollständige chirurgische Resektion die einzige Chance auf eine Heilung.
Phäochromozytom, Paragangliom
Etwa die Hälfte der Nebennierentumore sind hormonaktiv und können zu charakteristischen Begleitsymptomen führen. Vor jeder invasiven Maßnahme muss ein Phäochromozytom (Phäo) ausgeschlossen werden. Eine unzureichende Diagnostik kann im Falle einer Hormonaktivität des Nebennierentumors durch direkte Manipulationen eine ungehemmte Katecholaminfreisetzung zur Folge haben, was häufig zum Tode führen kann.1
Die Prävalenz des Phäo bei Patienten mit einer arteriellen Hypertonie liegt zwischen 0,1 und 0,6% mit einer jährlichen Inzidenz in der Allgemeinbevölkerung von einem Erwachsenen pro 100000 Einwohner.2
Geprägt wurde der Begriff Phäochromozytom im Jahre 1912 durch den Berliner Pathologen Ludwig Pick.3 Dieser beobachtete eine braune (griechisch phaios) Farbe (griechisch chroma) der adrenomedullären Tumorzellen (neulateinisch -zytus, -om) bei Kontakt mit Chromsalzen. Phäochromozytom ist eine Sonderbezeichnung für ein Paragangliom (PGL), das dem Nebennierenmark entstammt.
PGL sind überwiegend Katecholamin-produzierende Tumoren chromaffiner Zellen. Diese können sich aus sympathischen und parasympathischen Ganglien im Abdomen, Thorax, Kopf- und Halsbereich entwickeln. Phäos machen bis zu 86% der PGL aus, während die übrigen 14% extraadrenal vorkommen. Sehr selten sind dabei mediastinale Lokalisationen oder ein Auftreten in Rektum, Blase und Prostata.4,5
Katecholaminmetaboliten
Die drei Katecholamine Dopamin, Noradrenalin und Adrenalin werden in Abhängigkeit von Differenzierungsort und -grad von Phäos/PGL gebildet. Biochemisch werden die Katecholamine in chromaffinen Granula ATP-abhängig gespeichert und binden dabei an Chromogranin A.6 Innerhalb der chromaffinen Zellen werden die Katecholamine unabhängig von der Katecholaminsekretion kontinuierlich metabolisiert. Dabei entsteht aus Dopamin Methoxytyramin, aus Noradrenalin Normetanephrin und aus Adrenalin Metanephrin. Die jeweilige Sekretion gibt Hinweise auf die Tumorlokalisation (Tab. 1).
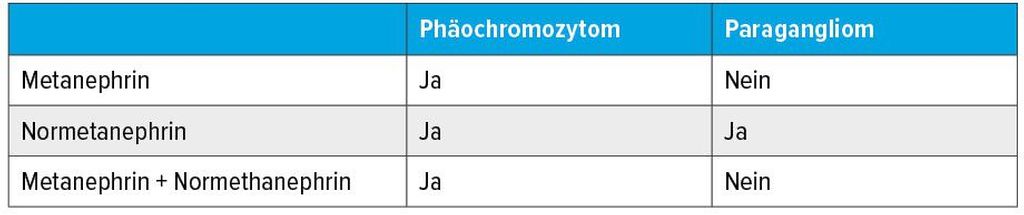
Tab. 1: Hinweise auf Tumorentität anhand der gemessenen Katecholaminmetaboliten
Erhöhtes Metanephrin spricht für eine Tumorlokalisation des Nebennierenmarks, da nur dort die Phenylethanolamin-N-Methyltransferase vorhanden ist und die Umwandlung von Noradrenalin in Adrenalin katalysiert wird. Phäos bilden etwa zu 50% der Fälle Noradrenalin, die andere Hälfte Noradrenalin und Adrenalin. Abdominelle PGL besitzen keine Phenylethanolamin-N-Methyltransferase und bilden daher überwiegend Noradrenalin und gelegentlich zusätzlich Dopamin. Rund 15–20% der PGL, v.a. die des Halsbereiches, sezernieren keine Katecholamine und sind asymptomatisch.
Genetik
Bei zumindest einem Drittel der Erkrankten mit Phäo/PGL liegt eine prädisponierende Genmutation vor.7 Patienten, die in einem Alter <45 Jahren erkranken, haben ein 5-fach erhöhtes Risiko einer Mutation gegenüber >45-Jährigen.8 Männer und Frauen mit genetischer Prädisposition erkranken gleich häufig, während bei sporadischen PGL mehr Frauen betroffen sind als Männer (1,5:1).
Unterschiedliche genetische Veränderungen führen zu spezifischen Phäo/PGL, weshalb anhand der Lokalisation des Tumors und des Katecholaminsekretionsprofils eine zielgerichtete genetische Testung durchgeführt werden kann.9
Mutationen in den Tumorsuppressorgenen SDHB, SDHC, SDHD sind am häufigsten zu finden. Eine SDHB-Mutation im Speziellen geht häufig mit Dopamin- oder Noradrenalin-sezernierenden extraadrenalen PGL einher, die oftmals Metastasen bilden.9 VHL-Genmutation-assozierte Phäos/PGL sezernieren ausschließlich Noradrenalin. Bilateral auftretende Phäos treten oft mit den Mutationen im RET-, VHL-, NF1-Gen auf, sind aber nicht wie SDHx-Syndrome mit multifokalen PGL assoziiert. Hereditäre PGL kommen häufiger an unterschiedlichen Lokalisationen vor. Etwa bei 25% der betroffenen Phäo-Patienten liegt ein autosomal dominant vererbtes Syndrom vor, wie das Von-Hippel-Lindau-Syndrom, die multiple endokrine Neoplasie (MEN-2) und die Neurofibromatose Typ 1 (Morbus Recklinghausen). Das autosomal dominant vererbte Paragangliom-Syndrom führt durch eine SDHx-Mutation zu PGL v.a. im Kopf-Hals-Bereich.
Klinik
Art, Ausmaß und Frequenz der Symptome eines Phäo/PGL sind von dem Sekretionsverhalten des Tumors abhängig und können somit sehr unspezifisch sein und intermittierend auftreten. Dabei kommt es v.a. bei Adrenalin- und/oder Noradrenalin-produzierenden Phäo/PGL zu Beschwerden wie Kopfschmerzen, Schweißausbrüchen, Herzrhythmusstörungen, Palpitationen, Zittern, Blässe, Übelkeit und Erbrechen.10 Die oft als typisch beschriebene Beschwerdetrias bestehend aus Kopfschmerzen, Schwitzen und Tachykardien ist mit 19% selten.11
Einflussfaktoren wie Stress, Medikamente (z.B. Amphetamine, Antidepressiva, Glukokortikoide), mechanische Manipulation des Tumors (Druck auf das Abdomen, Sport, Biopsie, Harnlassen bei PGL in der Harnblase) sowie Nahrungsmittel reichlich biogene Amine enthalten (Käse, Wein, Schokolade, Kaffee) können Symptome provozieren. Dopamin-ausschüttende PGL tendieren dazu, in erster Linie durch organverdrängende Wachstumsprozesse, wie Schmerzen, auffällig zu werden. Katecholamin-assoziierte Symptome sind hier sehr selten. Sie neigen häufiger zu Rezidiven und entwickeln häufiger Metastasen als PGL, die ausschließlich Adrenalin und Noradrenalin produzieren.9
Infolge der vermehrten Durchführung von Computertomografien (CT) oder Magnetresonanztomografien (MRT) zur Abklärung Phäo/PGL-unabhängiger Beschwerden werden in den letzten Jahren gehäuft Patienten mit präklinischer Erkrankung entdeckt. Etwa 60% der Diagnosen bei asymptomatischen Patienten sind mittlerweile inzidentell.12
Biochemische Diagnostik
Die Bestimmung der freien Metanephrine und Normetanephrine im Blut und Urin ist die sensitivste Methode zur Diagnostik der Phäos/PGL. Die Messung der freien Plasmametanephrine hat eine sehr hohe Sensitivität (99%), aber eine deutlich geringere Spezifität (85–90%). Um eine korrekte Diagnostik zu gewährleisten, ist das Untersuchungssetting ungemein wichtig. Zudem sind die Ergebnisse angepasst an die vom Patientenalter abhängigen Referenzwerte zu interpretieren.
Ein systematischer Review konnte zeigen, dass bei 97% der Patienten, die aufgrund einer arteriellen Hypertonie eine Metanephrin-Plasmabestimmung erhalten haben, trotz eines positiven Laborbefundes kein Phäo/PGL vorliegt.13 Diese gehäuft falsch-positive Diagnose eines vermeintlichen Phäo/PGL verursacht Probleme. So sollte die Blutabnahme am Morgen bei einem nüchternen Patienten erfolgen, der zuvor 20min ruhig auf dem Rücken lag. Störfaktoren sollten vermieden werden. Blutabnahmen im Sitzen führen zu einer 6-fach höheren Rate an falsch-positiven Ergebnissen. Zudem sollte ein venöser Zugang vor der Blutabnahme gelegt werden und Stressfaktoren und Zigarettenrauchen sollten vor der Blutabnahme unbedingt vermieden werden. Einige Medikamente interferieren mit den Testergebnissen und sollten vor der Analyse abgesetzt werden. Falsch-positive Ergebnisse treten z.B. nach Einnahme von Antidepressiva, Sympathomimetika, Alphablockern, Kalziumkanalblockern und Levodopa auf. Insgesamt brauchen die Untersuchungen ein etabliertes Setting und können nicht nebenbei erfolgen.
Bei der 24-h-Urin-Untersuchung der Katecholaminmetaboliten ist es wichtig, dass der Patient ausnahmslos eine exakte Harnsammlung durchführt. Hierbei kommt es oft zu Fehlern. PGL werden durch die Metabolitenbestimmung im Urin mit einer Sensitivität von 92,4% und einer Spezifität von 94,2% detektiert.
Weil Chromogranin A mit Katecholaminen der sekretorischen Granula kosezerniert, kann dieser Tumormarker ebenfalls im Blutserum oder -plasma bestimmt werden. Chromogranin A wird vorrangig in der Diagnostik und Nachsorge von PGL eingesetzt, die normwertige Harn- und/oder Plasmametanephrine aufweisen. Die Interpretation des Markers muss umsichtig erfolgen, da erhöhte Werte ebenfalls bei anderen neuroendokrinen Tumoren, einer Niereninsuffizienz, Herzerkrankungen und der Einnahme von Histaminblockern auftreten können.
Ein Clonidin-Suppressionstest wird bei grenzwertig isoliert erhöhten Katecholaminmetaboliten durchgeführt. Clonidin, als zentral wirksamer α2-Agonist, supprimiert die Exkretion von Katecholaminen. Im Fall eines paradoxen Anstiegs oder Ausbleibens eines signifikanten Absinkens des Ausgangswertes liegt eine autonome Katecholaminsekretion durch ein Phäo/PGL vor.
Bildgebung
Die CT und die MRT bieten eine weitgehend äquivalente diagnostische Genauigkeit für die bildgebende Abklärung eines Phäo. Bei der Detektion sporadischer Phäo wird eine Sensitivität von 98%, aber eine recht niedrige Spezifität von 70% angegeben.14 Das bedeutet, dass kaum Phäo in der Bildgebung übersehen werden, aber wenn etwas gesehen wird, muss dies mit weiterer Diagnostik spezifiziert werden. In der CT zeichnen sich Phäo nahezu ausnahmslos durch Dichtewerte ≥10HU aus und imponieren solide, teils gemischt-zystisch (Abb.1).15
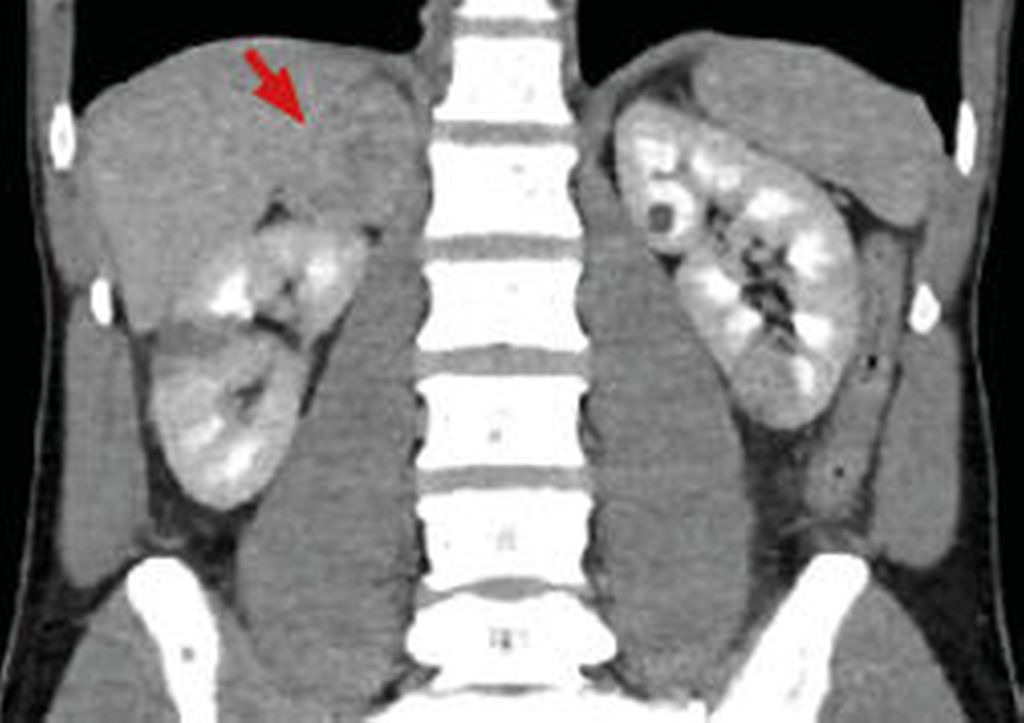
Abb. 1: Koronare Ansicht einer venösen Phase einer Abdomen-CT mit Nachweis eines Phäochromozytoms rechts (Pfeil). Nebenbefundlich Niereninfarkt mittleres Drittel rechts
Diese Informationen sind vor allem im Kontext von häufig gefundenen Inzidentalomen wichtig. Inzidentalome werden mittels Nativ-CT diagnostiziert. Sind diese homogen, ≤4cm, haben Fett als Inhalt oder HU-Einheiten ≤10, so sind diese gutartig und es bedarf keiner weiteren Bildgebung.16
Sowohl für die CT als auch für die MRT sind genaue Kriterien für die Abklärung von Nebennierentumoren definiert. In T2-gewichteten Bildern der MRT präsentieren sich Phäo im Vergleich zur Leber hyperintens und andere Nebennierentumoren isointens. Die Darstellung von intrazellulärem Fett mittels „Chemical shift“-Bildgebung schließt ein Phäo mit einer Sensitivität von 94% und einer Spezifität von 95% aus.17
In den letzten Jahren gewann die nuklearmedizinische Bildgebung zum Staging des Phäo/PGL immer mehr an Bedeutung. Die 123I-Metaiodobenzylguanidin(MIBG)-Szintigrafie, die 18F-Fluorodeoxyglukose-(FDG)-PET/CT, die 18F-Fluorodihydroxyphenylalanin(FDOPA)-PET/CT oder die Somatostatinrezeptor-PET/CT haben eine wesentlich höhere Spezifität als die konventionelle Bildgebung (CT/MRT). Dabei hängt die jeweilige Sensitivität der Tracer vom Mutationsstatus und der Tumorregion ab.18 Insbesondere die 18F-FDOPA-PET ist für die Darstellung von Gewebe mit Katecholaminmetabolismus geeignet. 18F-FDOPA wird über Aminosäuretransporter in die Zellen aufgenommen und in 18F-Dopamin metabolisiert, das sich in Katecholaminspeichervesikeln anreichert. Im Gegensatz zu 123I-Metaiodobenzylguanidin (123I-MIBG) reichert sich 18F-FDOPA in der Regel nicht in normalen Nebennieren an.18 Die Sensitivität von 18F-FDOPA liegt für sporadische Phäo mit 100% besonders hoch. Hereditäre Tumoren mit SDHB-Gen-Mutation werden allerdings nur mit einer Sensitivität von 20% detektiert.18 Für diese PGL und Metastasen eignen sich v.a. Bildgebungen zur Darstellung der Somatostatinrezeptoren. Hierzu werden in der PET-Diagnostik Somatostatinanaloga über einen Chelator wie DOTATOC, DOTANOC und DOTATATE mit verschiedenen trivalenten Radiometallen markiert, z.B. 68-Ga DOTATATE-PET.19
Die 18F-Fluorodeoxyglukose-Positronenemissionstomografie (FDG-PET oder FDG-PET/CT) ist ein weiteres im klinischen Alltag zum Einsatz kommendes nuklearmedizinisches Verfahren. Ähnlich wie Metastasen sind Phäo/PGL durch eine gesteigerte Aufnahme von 18F-FDG gekennzeichnet. FDG reichert sich bei malignen PGL stärker an als bei benignen PGL (Sensitivität: 88% vs. 58%), da sie einen erhöhten Glukoseverbrauch aufweisen. Ein präoperatives FDG-PET präoperativ sollte durchgeführt werden: bei PGL, bei Phäos mit erhöhten 3-Methoxytyramin-Level und bei SDH-Mutation.18
Therapie
Die operative vollständige Entfernung des lokal begrenzten Phäos/PGL ist die Therapie der Wahl. Um die Wirkung von klinisch relevanten Katecholamin-sezernierenden Phäo/PGL aufzuheben, wird zumindest 7 Tage vor einer operativen Entfernung eine medikamentöse Therapie eingeleitet. Hierbei ist eine unspezifische α-Rezeptorblockade durch Phenoxybenzamin oder spezifisch bindende α-Rezeptorblocker (z.B. Doxazosin, Prazosin und Terazosin) möglich.
Spezifische α-Antagonisten bergen durch ihre geringe Halbwertszeit das Risiko, intraoperativ hypertensive Krisen auszulösen. Die Gefahr einer postoperativen schweren Hypotension ist nach Gabe von Phenoxybenzamin allerdings höher. Die Medikamentendosierung erfolgt individuell und muss unter strengem Monitoring angepasst werden, bis ein adäquates Absinken des Blutdrucks erreicht wird. Nebenwirkungen wie eine Reflextachykardie können durch β-Blocker behandelt werden. Diese dürfen nur gemeinsam und nach Einleiten der α-Blockade eingenommen werden, da es sonst zu kardiopulmonaler Dekompensation und hypertensiven Krisen kommen kann als Resultat einer ungehinderten α-Adrenorezeptor-Stimulation.20
Einige Patienten profitieren von einer Therapie mit Kalziumantagonisten wie Amlodipin, die den Noradrenalin-vermittelten Kalziumeinstrom in die glatte Gefäßmuskulatur blockieren. Eingesetzt werden Kalziumantagonisten, wenn es unter α-Blockaden zu schweren Hypotonien kommt oder keine ausreichende Blutdruckkontrolle erreicht werden kann.21 Postoperativ ist neben dem Monitoring des Blutdrucks die Überwachung von Herzfrequenz und Glukosespiegel wichtig, insbesondere wenn eine (partielle) Adrenalektomie durchgeführt wurde. Kopf- und Hals-PGL der parasympathischen Ganglien oder biochemisch stille PGL benötigen keine α-Rezeptorblockade.
In ausgewählten Fällen ist auch vor OP eines Phäo ein Verzicht auf eine α-Blockade möglich. Dieses Vorgehen wird unterstützt durch eine Studie der Arbeitsgruppe um Groeben, welche 110 Patienten mit und 166 Patienten ohne perioperative α-Blockade verglich. Es zeigten sich keine Unterschiede in den beiden Gruppen hinsichtlich intraoperativen systolischen Blutdrucks, hypertensiver Krisen oder schwerer Komplikationen.22
Durch verbesserte Diagnostik mit genauer Lokalisation des Tumors und exakter minimal invasiver „No touch“-Operationstechnik ohne langfristige α-Blockade können z.B. Patienten mit vorbestehenden kardio- bzw. zerebrovaskulären Vorerkrankungen profitieren, die besonders empfindlich auf hypotensive Episoden reagieren, die regelhaft unter α-Blockade auftreten.20
Die oftmals verwendete Grenze der Tumorgröße zur minimal invasiven Entfernung des Phäo liegt bei 6cm. Eine laparoskopische trans- oder retroperitoneale Tumorentfernung ist durchaus auch bei Raumforderungen >6cm möglich.23,24 Vorteile des laparoskopischen gegenüber dem offenen Zugang sind eine kürzere Krankenhausaufenthaltsdauer, geringerer Transfusions- und postoperativer Analgesie-Bedarf. Der offen-chirurgische Standardzugang bei größeren Tumoren erfolgt über einen transabdominellen Subkostalschnitt und hat den Vorteil einer vollständigen Freilegung und Einsicht in den Situs.25 Ein Funktionserhalt durch eine subtotale Adrenalektomie sollte v.a. bei bilateralen Tumoren durchgeführt werden. Hierbei soll der Erhalt von mindestens einem Drittel einer Nebenniere erreicht werden. Dies stellt eine ausreichende Hormonproduktion sicher, wodurch eine lebenslange Glukokortikoidsubstitution und Nebenwirkungen wie Fatigue, Hypocortisolismus bis hin zum Tod vermieden werden kann.26
Insgesamt bedarf es auch bei der Therapie interdisziplinärer Erfahrung.
Histologie
Zwischen 8,3 und 13% der Phäo/PGL sind maligne.27 Laut WHO ist die Bezeichnung eines metastasierten anstelle eines malignen Phäo/PGL zu bevorzugen, da nur der Nachweis von Metastasen in sonst Chromaffinzellen-freien Regionen (Leber, Knochen, Lunge, Lymphknoten) ein Beweis eines malignen Phäo/PGL ist. Interessanterweise ist eine histologische Unterscheidung zwischen malignem und benignem Gewebe nicht möglich.20
Potenzielle Risikofaktoren einer Metastasierung sind große Tumoren >5cm, ein hoher Ki-67-Index >3%, SDHB-Mutationen, >3-fach erhöhte Plasma-Methoxytyramin-Werte und ein PASS (Pheochromocytoma of the Adrenal Gland Scaled Score) >6, der u.a. die Kapselinvasion, den Zellkernpleomorphismus und die Hyperchromasie des Zellkerns beurteilt.28
Eine weitere Risikostratifizierung ist mit dem GAPP-Score (Grading System for Adrenal Pheochromocytoma and Paraganglioma) möglich und Werte zwischen 7 und maximal 10 Punkten sprechen bei einem wenig differenzierten Tumor für eine Metastasierungswahrscheinlichkeit von >90%.
Jedes lokalisierte Phäo/PGL besitzt das Potenzial zu metastasieren. In der Literatur sind Fälle einer Metastasierung >50 Jahre nach ursprünglicher Lokaltherapie beschrieben, weshalb ein lebenslanges jährliches Follow-up für Patienten mit erhöhtem Metastasierungsrisiko empfohlen wird.27, 29 Dieses besteht aus der Bestimmung der Katecholaminmetaboliten und regelmäßiger Bildgebung. Eine aktuelle multizentrische Register-Studie, PROSPHEO (NCT03344016), untersucht das Outcome eines intensiven oder Standard-Follow-ups nach Operation.
Adrenokortikales Karzinom
Das Nebennierenkarzinom als maligne Entität der Nebennierenrinde ist ein sehr seltener und hochmaligner Tumor, dessen einzige kurative Therapie aus der vollständigen operativen Entfernung besteht. Weltweit tritt das adrenokortikale Karzinom (ACC) mit einer Inzidenz von 0,2–2 pro 1 Million pro Jahr auf. Männer und Frauen sind in einem Verhältnis von 1,5:1 betroffen. Es existieren zwei Erkrankungsgipfel, zum einen im Kindesalter vor dem 10. Lebensjahr, zum amderen in der 5. und 6. Lebensdekade, wobei die Erkrankung im Erwachsenenalter aggressiver und letaler verläuft.30
Der Tumor ist in den meisten Fällen zunächst asymptomatisch und wird zufällig aufgrund einer CT oder MRT, die Nebennieren-unabhängig durchgeführt wird, entdeckt. Ab einer bestimmten Größe kommt es zur Verdrängung umliegenden Gewebes oder zu Organinfiltrationen, welche zu Flankenschmerzen, Übelkeit und Erbrechen sowie Verdauungsstörungen führen können.31 Bei der Entdeckung weist der Tumor im Median schon einen Durchmesser von >11cm auf, wogegen benigne Nebennierentumoren oft <5cm sind.32
Etwa 40% der ACC sind hormoninaktiv, während 60% der ACC autonom Hormone produzieren, die in unterschiedlicher, teils präklinischer Ausprägung zu Begleitsymptomen der Hormonüberproduktion führen.31 Knapp die Hälfte der Patienten zeigt ein Cushing-Syndrom, etwa 25% weisen ein gemischtes Cushing- und Virilisationssyndrom auf, das durch eine Überproduktion von Glukokortikoiden und Androgenen verursacht wird. Bei etwa 10% treten ausschließlich Virilisierungszeichen auf.31
Hormonanalyse beim ACC
Die primäre Hormondiagnostik dient dazu, den Verdacht eines malignen ACC zu erhärten und das Hormonexkretionsprofil einzuordnen. Ein massiver Überschuss an adrenokortikalen Steroiden kann die Lebensqualität und Überlebenszeit reduzieren, sodass eine Hormonexkretionsblockade notwendig wird. Nach Identifikation der genauen Hormonsekretion wird dies zudem als Tumormarker zur Verlaufskontrolle genutzt.16, 33
Bis zu 95% der Patienten mit autonomer Hormonsekretion des ACC haben keine oder nur subtile Symptome.34 Im Falle eines subklinisch autonomen Steroidexzesses kommt es nach einer Adrenalektomie zu einer behandlungsbedürftigen sekundären Nebenniereninsuffizienz, die präoperativ durch die Bestimmung des Kortikotropin-Releasing-Hormons quantifiziert werden kann.
Somit wird bei ACC-Verdacht auf eine Überproduktion der Sexualsteroide bzw. Steroidvorstufen (DHEA-S, 17-OH-Progesteron, Androstendion, Testosteron) sowie der Glukokortikoide (Dexamethasonhemmtest, freies Kortisol im 24h-Urin, basales Plasma-ACTH, basales Serumkortisol) und Mineralokortikoide (Kalium, Aldosteron-Renin-Ratio) getestet. Differenzialdiagnostisch ist es obligat, durch Messung der Katecholaminmetaboliten ein Phäo auszuschließen. Gerade hier ist die Zusammenarbeit mit Endokrinologen wichtig.
Bildgebung
Bei der primären Bildgebung ist zwingend eine CT des Abdomens und des Thorax notwendig, idealerweise nativ und mit einem 10-min-Kontrastmittel-Washout (Abb. 2). Die meisten ACC sind inhomogen, zeigen kein Washout in der Spätphase und sind fettarm (HU >10).33 Alternativ ist eine MRT möglich. Die 18F-FDG-PET zeigte in einer Metaanalyse eine hohe Sensitivität von 97% und eine Spezifität von 91% zur Unterscheidung benigner von malignen Nebennierentumoren. Eine Läsion gilt dann als benigne, wenn das Uptake dem in der Leber entspricht oder geringer ausfällt.35
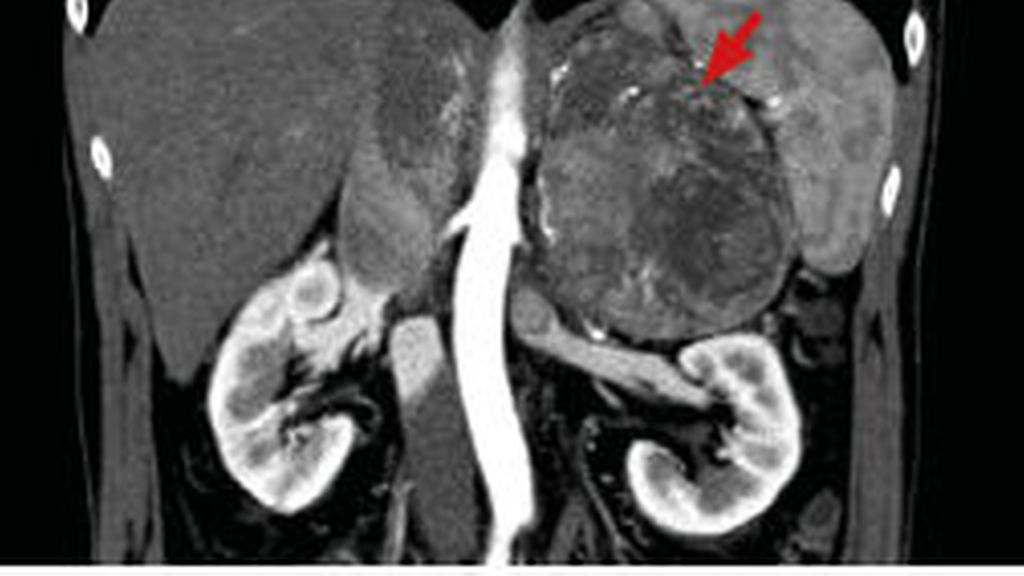
Abb. 2: Axiale Ansicht einer CT eines ausgedehnten, teils nekrotischen Nebennierenkarzinoms mit Infiltration der V. cava inferior und V. renalis links (Pfeil)
Biopsie
Bei Verdacht auf ein ACC ist eine wichtige Frage, ob eine Biopsie durchgeführt werden sollte. Die Antwort darauf ist bis auf wenige Ausnahmen „Nein“. Dies gilt insbesondere dann, wenn eine chirurgische Resektion möglich ist.
Die histopathologische Differenzierung zwischen ACC und anderen Tumorentitäten ist unsicher; die Genauigkeit beträgt 42–70%.36 In bis zu 11% kommt es infolge einer Biopsie zu Blutungen. Bedenken, dass es zu einer Tumoraussaat im Retroperitoneum und im Stichkanal kommen kann, sind eher zu vernachlässigen, aber in 1,3% der Fälle möglich.36
Ausnahmen gibt es im Falle eines inoperablen Tumors, wie in Abbildung 2 dargestellt, um eine systemische medikamentöse Therapie zu planen oder wenn der Verdacht besteht, dass es sich um eine Metastase eines extraadrenalen Malignoms handeln könnte und eine therapeutische Konsequenz daraus zu erwarten ist.37
Die TNM-Klassifikation des Nebennierenkarzinoms wurde 2017 überarbeitet. Das T1-Stadium beschreibt einen ≤5cm großen, das T2-Stadium einen >5cm großen Nebennierentumor, jeweils ohne extraadrenales Wachstum. Im T3-Stadium wächst der Tumor lokal invasiv, aber ohne umgebende Organe zu infiltrieren. Im Falle eines T4-Tumors werden umliegende Organe oder Gefäße vom Tumor infiltriert, wie Niere, Diaphragma, Leber, A. renalis oder Vena cava. In Kombination mit der Klassifikation des European Network for the Study of Adrenal Tumors (ENSAT) können 4 Stadien definiert werden, mit der das krebsspezifische 5-Jahres-Überleben (CSS) angegeben werden kann.38
In den nicht fernmetastasierten StadienI (T1N0M0) und II (T2N0M0) liegt das 5-Jahres-CSS jeweils bei 82 und 61%. Im Stadium III (T3 oder T4N1M0) liegen bereits regionale Lymphknoten- aber keine Fernmetastasen vor. Etwa 50% der Patienten mit dieser Konstellation leben nach 5 Jahren. Liegen Fernmetastasen vor, ist die Prognose infaust. Im Stadium IV (M1) liegt das 5-Jahres-CSS bei 13%. Metastasen finden sich in erster Linie in Lunge (47–97%) und Leber (53–68%), gefolgt von Knochen (11–33%).
Operative Therapie des ACC
In Stadium I–II wird eine operative Entfernung in einem spezialisierten Zentrum empfohlen. Oberstes Ziel ist die komplette Entfernung des Tumors (R0-Resektion) ohne Verletzung der Tumorkapsel sowie eine lokoregionale Lymphadenektomie. Beim ACC ist die offene Operation Standard. Im Falle eines ACC <6cm ohne Hinweise einer lokalen Invasion ist eine laparoskopische Adrenalektomie möglich, wenn eine R0 sichergestellt werden kann.39
Die histologische Beurteilung der adrenalen Neoplasie sollte von einem erfahrenen Pathologen durchgeführt werden. Mehr als drei erfüllte Kriterien im Weiss-Score (Kernatypien, atypische Mitosen, >5 Mitosen pro 50 High-Power Field, <25% klarzellige Tumorzellen, >33% diffuse Zellarchitektur, Nekrosen, Veneninvasion, Sinusinvasion, Kapselinvasion) und ein Ki-67-Index >10% als Zeichen vermehrter Zellproliferation sind typisch für ein ACC.33, 39 Eine adjuvante Therapie mit Mitotan ist bei hohem Rezidivrisiko im lokalisierten Stadium I, II und R0-Resektion ab einem Ki-67-Wert >10% zu diskutieren. Diese sollte mindestens 2 Jahre und maximal 5 Jahre erfolgen.
Auch in den höheren Stadien (III, IV) ist die chirurgische Entfernung die einzige Heilungschance. Neoadjuvante Studien laufen. Auch hier ist es wichtig, eine R0-Resektion anzustreben. Hier ist immer eine adjuvante Mitotan-Therapie einzuleiten. Bei einer R1- oder Rx-Resektion ist eine adjuvante Radiatio (50–60Gy) zu besprechen. Bei initial metastasierten adrenokortikalen Karzinomen oder Rezidiven werden in erster Linie systemische Therapien eingesetzt.
Literatur:
1 Arellano RS et al.: Image-guided percutaneous biopsy of the adrenal gland: review of indications, technique, and complications. Curr Probl Diagn Radiol 2003; 32(1): 3-10 2 Kolačkov K et al.: Genetic aspects of pheochromocytoma. Adv Clin Exp Med 2012; 21(6): 821-9 3 Pick L: Das ganglioma emblionale sympathicum. Berl Klin Wschr 1912; 49: 16-22 4 Pamporaki C et al.: Characteristics of pediatric vs adult pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 2017; 102(4): 1122-32 5 Farrugia F-A, Charalampopoulos A: Pheochromocytoma. Endocr Regul 2019; 53(3): 191-212 6 Johnson RG et al.: Proton: substrate stoichiometries during active transport of biogenic amines in chromaffin ghosts. J Biol Chem 1981; 256(11): 5773-80 7 Galan SR, Kann PH: Genetics and molecular pathogenesis of pheochromocytoma and paraganglioma. Clin Endocrinol (Oxf) 2013; 78(2): 165-75 8 Erlic Z et al.: Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res 2009; 15(20): 6378-85 9 Foo SH et al.: Dopamine-secreting phaeochromocytomas and paragangliomas: clinical features and management. Singapore Med J 2010; 51(5): e89-93 10 Lenders JWM et al.: Phaeochromocytoma. Lancet 2005; 366(9486): 665-75 11 Geroula A et al.: Pheochromocytoma and paraganglioma: clinical feature-based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. Eur J Endocrinol 2019; 181(4): 409-20 12 Oshmyansky AR et al.: Serendipity in the diagnosis of pheochromocytoma. J Comput Assist Tomogr 2013; 37(5): 820-3 13 Sawka AM et al.: A systematic review of the literature examining the diagnostic efficacy of measurement of fractionated plasma free metanephrines in the biochemical diagnosis of pheochromocytoma. BMC Endocr Disord 2004; 4(1): 2 14 Ilias I, Pacak K: Diagnosis, localization and treatment of pheochromocytoma in MEN 2 syndrome. Endocr Regul 2009; 43(2): 89-93 15 Buitenwerf E et al.: Unenhanced CT imaging is highly sensitive to exclude pheochromocytoma: a multicenter study. Eur J Endocrinol 2018; 178(5): 431-7 16 Fassnacht M et al.: Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016; 175(2): G1-G34 17 Raja A et al.: Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201(4): 825-33 18 Taïeb D et al.: European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2019; 46(10): 2112-37 19 Deppen SA et al.: Safety and efficacy of 68Ga-DOTATATE PET/CT for diagnosis, staging, and treatment management of neuroendocrine tumors. J Nucl Med 2016; 57(5): 708-14 20 Neumann HPH et al.: Pheochromocytoma and paraganglioma. N Engl J Med 2019; 381(6): 552-65 21 Lenders JWM et al.: Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99(6): 1915-42 22 Groeben H et al.: Perioperative α-receptor blockade in phaeochromocytoma surgery: an observational case series. Br J Anaesth 2017; 118(2): 182-9 23 Palazzo FF et al.: Long-term outcome following laparoscopic adrenalectomy for large solid adrenal cortex tumors. World J Surg 2006; 30(5): 893-8 24 Walz MK: S2k Leitlinie Operative Therapie der Nebennierentumoren 2019 [Stand: 16.1.2021]. Verfügbar unter: https://www.awmf.org/uploads/tx_szleitlinien/088-008l_S2k_Operative-Therapie_Nebennierentumoren_2019-07.pdf 25 Hanna NN, Kenady DE: Pheochromocytoma. In: Hanna NN, Kenady DE (Hrsg.): Surgical Treatment: Evidence-Based and Problem-Oriented. München: Zuckschwerdt, 2001 Verfügbar unter: https://www.ncbi.nlm.nih.gov/books/NBK7002/ 26 Alesina PF et al.: Minimally invasive cortical-sparing surgery for bilateral pheochromocytomas. Langenbecks Arch Surg 2012; 397(2): 233-8 27 Hamidi O et al.: Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab 2017; 102(9): 3296-305 28 Parenti G et al.: Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. J Oncol 2012; 2012: 872713 29 Plouin PF et al.: European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016; 174(5): G1-G10 30 Ng L, Libertino JM: Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol 2003; 169(1): 5-11 31 Funder JW et al.: The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016; 101(5): 1889-916 32 Fassnacht M, Allolio B: What is the best approach to an apparently nonmetastatic adrenocortical carcinoma? Clin Endocrinol (Oxf) 2010; 73(5): 561-5 33 Gaujoux S, Mihai R: European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma. Br J Surg 2017; 104(4): 358-76 34 Arlt W: Adrenal insufficiency. Clin Med (Lond) 2008; 8(2): 211-5 35 Boland GWL et al.: Characterization of adrenal masses by using FDG PET: a systematic review and meta-analysis of diagnostic test performance. Radiology 2011; 259(1): 117-26 36 Williams AR et al.: Transcutaneous biopsy of adrenocortical carcinoma is rarely helpful in diagnosis, potentially harmful, but does not affect patient outcome. Eur J Endocrinol 2014; 170(6): 829-35 37 Fassnacht M et al.: Update in adrenocortical carcinoma. J Clin Endocrinol Metab 2013; 98(12): 4551-64 38 Fassnacht M et al.: Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer 2009; 115(2): 243-50 39 Fassnacht M et al.: Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2020; 31(11): 1476-90
Das könnte Sie auch interessieren:
.jpg)
Erstes „Da Vinci Single Port“-OP-System in Österreich
Seit März 2025 werden an der Klinik Oberwart robotisch assistierte Eingriffe mit dem „Da Vinci Single Port“(SP)-Operationssystem durchgeführt. Prim. Dr. Gottfried Pfleger berichtet über ...

Heutiger Stellenwert der ESWL: Renaissance in Sicht?
Die ESWL (extrakorporale Stoßwellenlithotrypsie) ist eine Technik zur urologischen Steintherapie, die in den 80er-Jahren des vorigen Jahrhunderts in den klinischen Alltag Einzug gehalten ...

GLP-1-Therapien: Auswirkungen unter der Gürtellinie?
GLP-1-Rezeptoragonisten wie Semaglutid werden häufig zur Gewichtsreduktion eingesetzt. Mit der stark steigenden Anwendung rücken nun auch bislang wenig untersuchte Auswirkungen auf ...