
Das VEXAS-Syndrom
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
2020 wurde eine neue Erkrankung beschrieben: das VEXAS-Syndrom. Es handelt sich dabei um eine autoinflammatorische Systemerkrankung, die häufig mit einer Beteiligung der Lunge einhergeht.
Das VEXAS-Syndrom ist eine 2020 erstmals beschriebene autoinflammatorische Systemerkrankung, wobei VEXAS für „vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic“ steht.1, 2 Die zugrundeliegende Ursache ist eine erworbene somatische, pathogene Mosaikmutation am UBA1-Gen, das am X-Chromosom positioniert ist und für das E1-Enzym kodiert. Das E1-Enzym ist für die Ubiquitinierung von Proteinen verantwortlich und dient hauptsächlich zum Abbau von Molekülen. Die Erstbeschreibung erfolgte durch Beck DB et al. im „New England Journal of Medicine“, die das Syndrom bei 25 Männern identifizieren konnten.2 Mittels Transkriptionsanalysen konnten sie zeigen, dass die myeloische Zellreihe der Betroffenen eine verringerte Ubiquitinierung aufwies, wodurch es zu einer Überregulierung von proinflammatorischen Zytokinen wie IL-1, IL-6, IL-8, IFN-γ und TNF-α kommt.1, 2
Das klinische Bild
Das VEXAS-Syndrom betrifft aufgrund seiner Lage am X-Chromosom fast ausnahmslos Männer (in sehr seltenen Fällen kann die Erkrankung auch bei Frauen auftreten) und manifestiert sich vorwiegend im fortgeschrittenen Alter (das mediane Manifestationsalter liegt bei 64 Jahren). Gekennzeichnet durch rezidivierende Fieberschübe und deutlich erhöhte Entzündungszeichen weisen die Patienten eine Vielzahl an klinischen Symptomen auf, wobei Überlappungen mit verschiedenen rheumatologischen, dermatologischen und auch hämatologischen Erkrankungen bestehen.2 Insbesondere wird über das Auftreten einer rezidivierenden Polychondritis berichtet. Makrozytäre hyperchrome Anämie und Thrombozytopenie sind ebenfalls charakteristisch, häufig findet sich auch das Vollbild eines myelodysplastischen Syndroms.1–4 Auch dermatologische Veränderungen treten bei einem Großteil der Betroffenen auf, wie zum Beispiel eine neutrophile Dermatitis, schmerzhafte Papeln, Erythema nodosum, eine kutane Vaskulitis der kleinen und mittleren Gefäße sowie febrile Dermatose (Sweet-Syndrom).2, 5, 6 Aber auch Serositis, entzündliche Erkrankungen der Augen wie Uveitis und Episkleritis sowie Thrombosen, Muskelschmerzen und Arthritis können infolge des Syndroms auftreten. Bei einer Vielzahl der Patienten finden sich zudem pulmonale Konsolidierungen mit Alveolitis (49%) und respiratorischer Insuffizienz, bei 40% treten thromboembolische Komplikationen auf.1, 7
Tabelle 1 zeigt eine Übersicht über die Charakteristika, die die VEXAS-Kohorte von Beck DB et al. aufwies.2
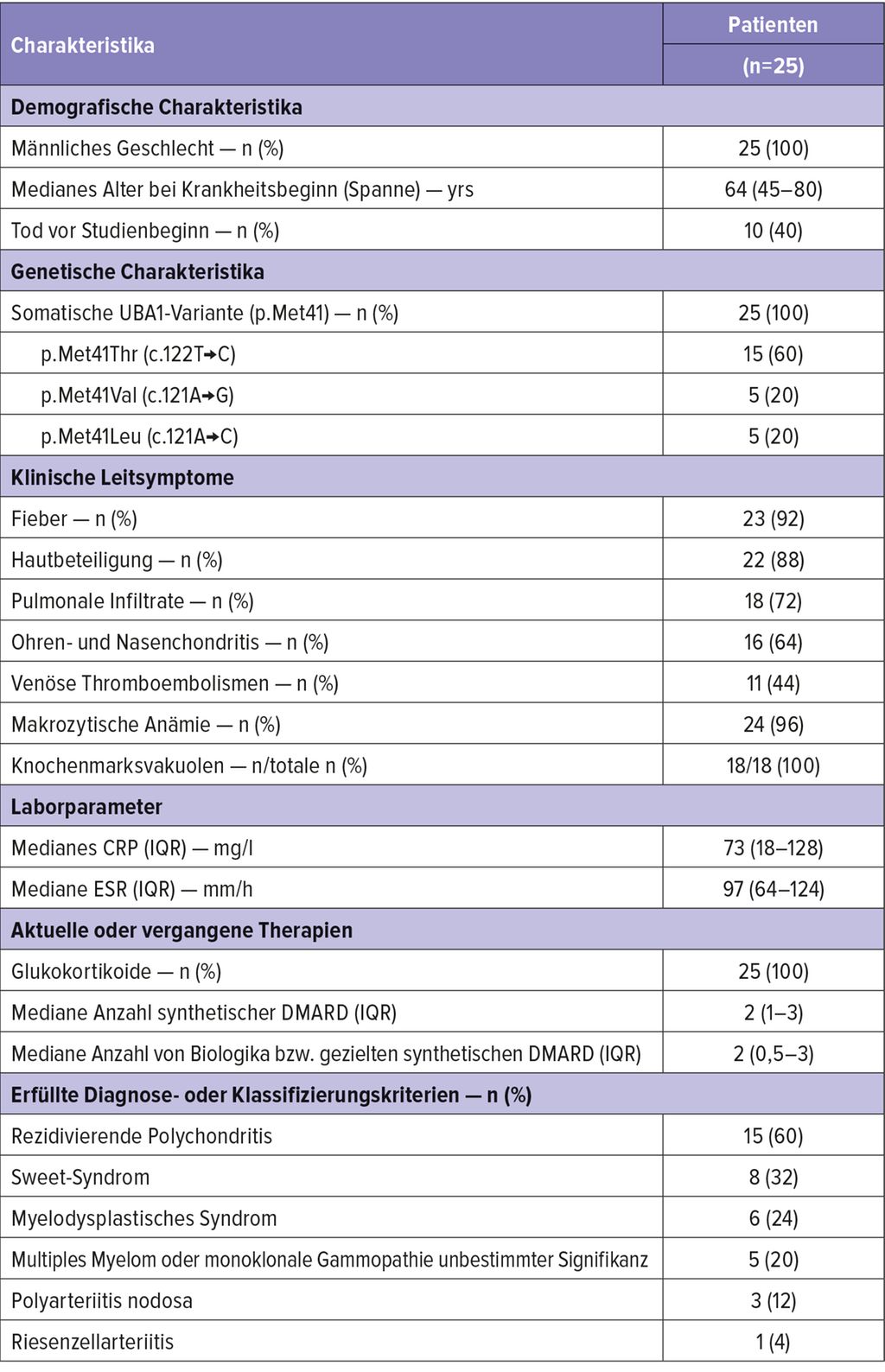
Tab. 1: Demografische und klinische Charakteristika der VEXAS-Kohorte von Beck DB et al. (modifiziert nach Beck DB et al. 2020)1
Diagnostik
Die serologischen Entzündungsparameter sind bei Patienten mit VEXAS-Syndrom meist stark erhöht. Blutbildveränderungen im Sinne einer makrozytären Anämie sind zu beobachten sowie eine Thrombopenie. In der Knochenmarkbiopsie lassen sich die namensgebenden zytoplasmatischen Vakuolen finden.1, 2, 8
Die genetische Testung auf die somatische Mutation des X-chromosomalen UBA1-Gens sichert die klinische Diagnose, es sollte jedoch bedacht werden, dass in der Sanger-Diagnostik Mosaike teilweise übersehen werden können und die diagnostische Sicherheit mittels Next-Generation-Sequencing erhöht werden kann.1
Therapie
Die Erkrankung ist meist mit sehr schwerwiegenden Komplikationen vergesellschaftet und geht mit einer hohen Mortalität einher – in der Kohorte von Beck DB et al. verstarben 40% der Patienten.2 Immunsuppression und Antiinflammation sind die Therapiekonzepte der Wahl: Neben verschiedenen antiinflammatorischen Therapieoptionen (z.B. Blockade von IL-1, IL-6 oder JAK) werden meist hohe Glukokortikoiddosen benötigt, um die Erkrankung zu kontrollieren.1
ÖGP-Awarenesskampagnen für seltene Erkrankungen
Auf Initiative der ÖGP-Expert:innengruppe „Interstitielle Lungenerkrankungen und Orphan Diseases“ wurde eine Awarenesskampagne für seltene Erkrankungen ins Leben gerufen, die jedes Jahr eine andere „orphan disease“ mit Lungenbeteiligung in den Fokus stellt. Im Zuge der heurigen ÖGP-Awarenesskampagne wurde für die ersten fünf Personen, die die Entdeckung eines VEXAS-Syndroms an die ÖGP melden, eine freie Registrierung für die ÖGP-Jahrestagung (23.–25.10. in Graz) ausgelobt.
Kontakt:
Einreichung der Diagnose eines VEXAS-Syndroms
Österreichische Gesellschaft für Pneumologie
OA Dr. Klaus Hackner
Leiter der ÖGP-Expert:innengruppe „Interstitielle Lungenerkrankungen und Orphan Diseases“
c/o Mondial Congress & Events
Tel.: +43158804-116
E-Mail:
ogp@mondial-congress.com
Quelle:
Presseaussendung der ÖGP vom 25.7.2023
Literatur:
1 Zeek M et al.: VEXAS-Syndrom. Z Rheumatol 2022; 81(9): 782-6 2 Beck DB et al.: Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2020; 383(27): 2628-38 3 Ferrada MA et al.: Somatic mutations in UBA1 define a distinct subset of relapsing polychondritis patients with VEXAS syndrome. Arthritis Rheumatol 2021; 73(10): 1886-95 4 Tsuchida N et al.: Pathogenic UBA1 variants associated with VEXAS syndrome in Japanese patients with relapsing polychondritis. Ann Rheum Dis 2021; 80(8): 1057-61 5 Zakine E et al.: UBA1 variations in neutrophilic dermatosis skin lesions of patients with VEXAS syndrome. JAMA Dermatol 2021; 157(11): 1349-54 6 Dehghan N et al.: Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome: fevers, myalgia, arthralgia, auricular chondritis, and erythema nodosum. Lancet 2021; 398(10300): 621 7 Shaukat F et al.: UBA1 and DNMT3A mutations in VEXAS syndrome. A case report and literature review. Mod Rheumatol Case Rep 2021; 6(1): 134-9 8 Gurnari C et al.: Vacuolization of hematopoietic precursors: an enigma with multiple etiologies. Blood 2021; 137(26): 3685-9
Das könnte Sie auch interessieren:


Infektion und Inflammation bei zystischer Fibrose
Die Lebenserwartung bei zystischer Fibrose hat sich dank großer therapeutischer Fortschritte gebessert, hochwirksame Kombinationstherapien mit CFTR-Modulatoren können Exazerbationen ...

Erklärungsversuch des Etagenwechsels von der Rhinitis zum allergischen Asthma
Der „Etagenwechsel“ von der allergischen Rhinitis zum Asthma ist ein zentrales Konzept in der Allergologie – und zugleich komplexer, als der Begriff vermuten lässt. Nach wie vor sind ...