Lungenbeteiligung bei systemischer Sklerose
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die systemische Sklerose (SSc) ist eine rheumatologische Erkrankung noch ungeklärter Ursache. Durch die drei Pathomechanismen der Entzündung, der Fibrose und der Vaskulopathie und durch den potenziellen Befall unterschiedlichster Organe kann die SSc ein sehr heterogenes Erscheinungsbild hervorrufen.
Keypoints
-
Negative ANA und negative SSc-Antikörper schließen eine SSc nicht aus.
-
Eine negative Kapillarmikroskopie lässt eine SSc ebenfalls nicht ausschließen.
-
Der Verlauf einer ILD kann schwer vorhergesagt werden, was eine Risikoeinstufung nötig macht.
-
Die Lungenfibrose kann eine Erstmanifestation einer SSc sein.
-
Regelmäßiges Screening bei SSc-Patienten ist unbedingt erforderlich.
-
Eine interdisziplinäre Zusammenarbeit zwischen Rheumatologie, Pulmologie und Radiologie im Sinne des ILD-Boards ist in jedem Fall angezeigt.
Die systemische Sklerose (SSc) ist eine seltene Erkrankung. Frauen sind dreimal häufiger betroffen. Der Erkrankungsgipfel liegt zwischen dem 50. und 70. Lebensjahr. In ca. 50% der SSc-Fälle kommt es zu einer Lungenbeteiligung im Sinne einer ILD („interstitial lung disease“).
Die Mortalität ist ebenso deutlich erhöht (2,3–3,5-fach). Risikofaktoren für eine erhöhte Sterblichkeit sind ein höheres Alter, das männliche Geschlecht, Proteinurie, erhöhte Entzündungswerte und eine verminderte Diffusionskapazität in der Lungenfunktionsmessung (DLCO).
Früher war die erhöhte Mortalität v.a. durch die renale Krise bedingt, mittlerweile lässt diese sichv.a. durch die Lungenfibrose, die pulmonal-arterielle Hypertonie (PAH), durch Malignome, kardiale Manifestationen oder Komplikationen und Infektionen erklären.
Typisch für die SSc ist v.a. der Hautbefall im Sinne einer verdickten Haut, eine Raynaud-Symptomatik, die im Verlauf zu digitalen Ulzera führen kann, sowie eine Lungenbeteiligung durch eine interstitielle Fibrose. Aber auch andere Organe, wie die Niere (renale Krise), das Herz (Rhythmusstörungen, Kardiomyopathie), der Gastrointestinaltrakt (gastroösophagealer Reflux, bakterielle Überbesiedelung des Darms, Malnutrition) und das muskuloskelettale System (Arthritis, Kalzinose) können betroffen sein.
Klassifikationskriterien
In die Klassifikationskriterien des/derACR/EULAR (American College of Rheumatology/European League Against Rheumatism) aus dem Jahr 2013 fließendie Hautverdickung, die Raynaud-Symptomatik, digitale Ulzera, Teleangiektasien, die pulmonale Hypertension, eine Lungenbeteiligung,die Kapillarmikroskopie und spezifische Autoantikörper mit ein. Aus der Gewichtung der Kriterien ergibt sich auch die Möglichkeit, eine SSc ohne das Vorhandensein von Antikörpern zu diagnostizieren. Im Umkehrschluss ist eine SSc bei negativenAutoantikörpern im Laborbefund nicht auszuschließen. Das ist auch ein Punkt, der in der Differenzialdiagnostik von Lungenfibrosen eine Rolle spielen sollte.
Autoantikörper
Daten der EUSTAR-Kohorte zeigten, dass ca. 93% der diagnostizierten SSc-Patienten ANA-positiv waren. 37% hatten Anti-Zentromer-Antikörper (Ak), 33% Anti-Topoisomerase-Ak (Scl-70) und 4,5% RNA-Polymerase-3-Ak. Daraus ergibt sich, dass 25% keine SSc-spezifischenAk aufwiesen. Als seltene Ak gelten Anti-Fibrillarin- (U3RNP), Anti-Th/To-, Anti-Pm-Scl-, Anti-U1RNP- und Anti-Ku-Ak. Auch aus diesen Daten ergibt sich, dass einerseits bei fehlenden ANA (ca. 7%) oder fehlenden gängigen SSc-Antikörpern eine SSc nicht ausgeschlossen werden sollte.
Je nach Antikörperstatus lässt sich das Risiko für den Krankheitsverlauf und eine bestimmte Organmanifestation einschätzen, daraus ergibt sich auch die Entscheidung für eineStrategie der Betreuung der Patienten. Anti-Zentromer-Ak scheinen v.a. das Risiko, eine PAH zu entwickeln, zu erhöhen. Anti-Topoisomerase-Ak (Scl-70-Ak) hingegen erhöhen das Risiko für eine interstitielle Lungenerkrankung im Rahmen einer SSc.
Aufgrund des potenziellen multiplen Organbefalls durch die SSc ist es wichtig, je nach Risikoprofil, das v.a. durch den Ak-Status bedingt ist, ein regelmäßiges Organscreening, v.a. auch im Rahmen der Erstdiagnostik, durchzuführen. So sollte jedenfalls als Basisdiagnostik eine Echokardiografie, Kapillarmikroskopie, Lungenfunktion (inkl. DLCO) und ein High-Resolution(HR)-CT des Thorax durchgeführt werden. Optionale Untersuchungen sind je nach Symptomatik und suspiziertem Organbefall durchzuführen.
Kapillarmikroskopie bei SSc
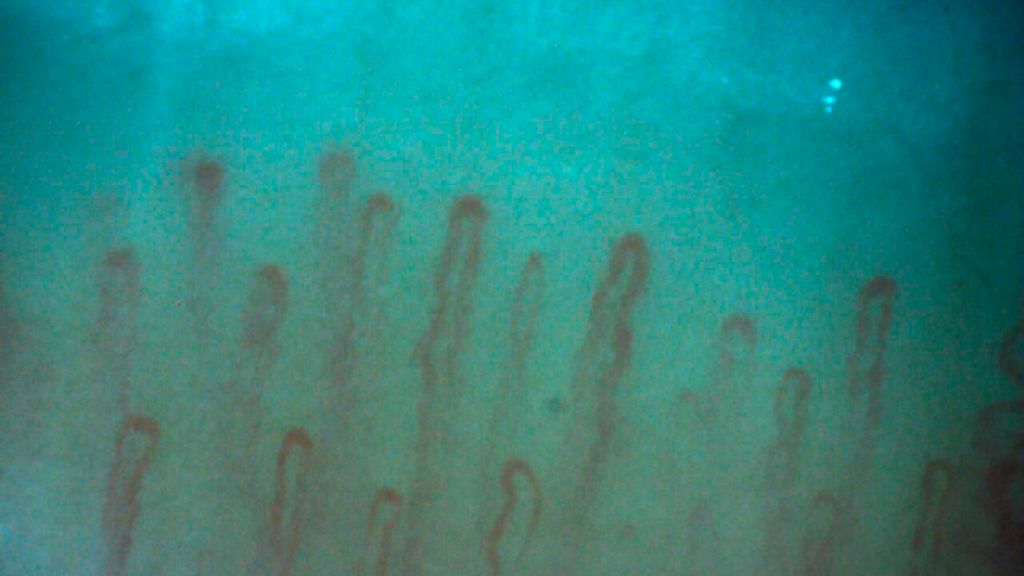
Die Kapillarmikroskopie ist ein wichtiges Tool zur Differenzierung, ob eine Raynaud-Symptomatik primär (d.h. ohne zugrunde liegende Erkrankung) oder sekundär (im Rahmen einer SSc) auftritt. In dieser an sich simplen Untersuchung, die auch in den Diagnosekriterien für die SSc verankert ist, können die Kapillaren sehr gut beurteilt werden. Hinsichtlich einer möglichen SSc ist ein frühes, ein aktives und ein Spätbild möglich. Differenziert wird anhand der Morphologie der Kapillaren, der Dichte und Ausrichtung und eventuellanhand von Mikroblutungen (Abb. 1 u. 2). In einer rezenten Studie wurden Auffälligkeiten in der Kapillarmikroskopie bei Patienten mit Lungenfibrose unklarer Genese untersucht, was die Frage aufwirft, ob Patienten mit Lungenfibrose zumindest im Rahmen der Basisdiagnostik häufiger zur Kapillarmikroskopie geschickt oder den Rheumatologen vorgestellt werden sollten. Vorsicht sei auch geboten, da in einer Studie gezeigt wurde, dass ca. 20% aller als primärer Raynaud klassifizierten Krankheitsbilder nach ca. 10 Jahren als sekundär eingeordnet wurden. Somit sollte auch bei einer unauffälligen Kapillarmikroskopie eine SSc nicht gänzlich ausgeschlossen werden.
©
B. Lindner
Abb. 1: Kapillarmikroskopie: normaler Befund | 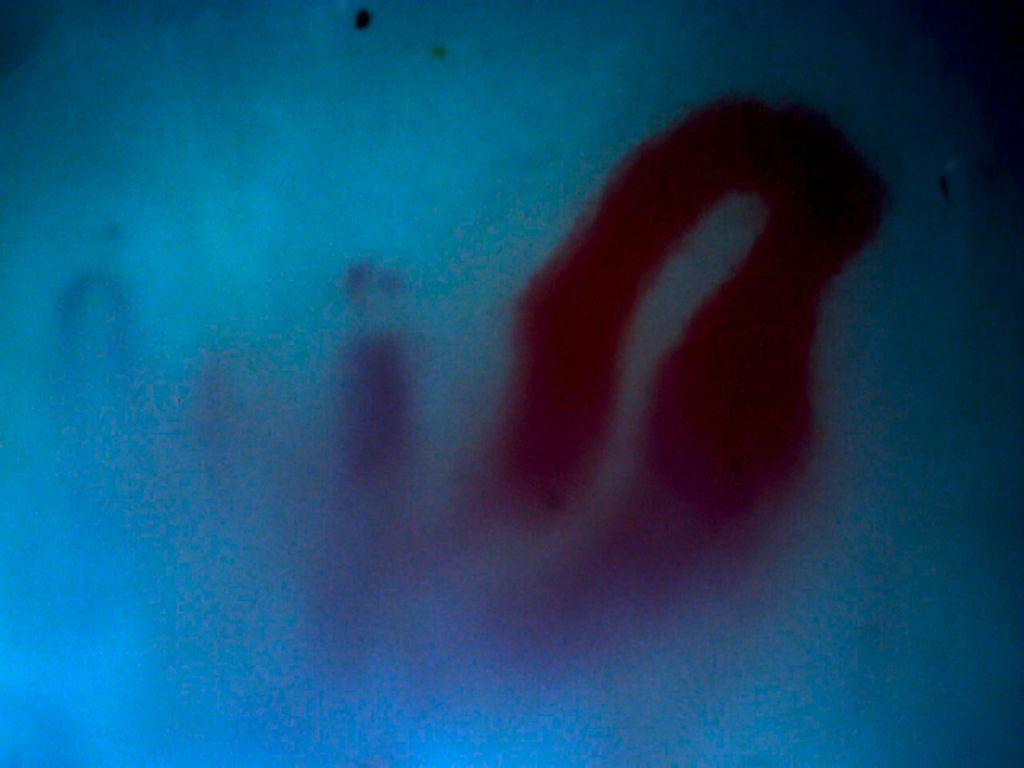
©
B. Lindner
Abb. 2: Kapillarmikroskopie: aktives Stadium einer SSc mit Megakapillare |
Differenzialdiagnose Dyspnoe bei SSc
Im Folgenden soll die respiratorische Situation der SSc-Patienten mit dem Hauptaugenmerk auf die Lungenfibrose behandelt werden.
Primär sei vorausgeschickt, dass nicht jeder Patient mit SSc, der an Dyspnoe leidet, eine Lungenfibrose haben muss. Häufige Differenzialdiagnosen im Rahmen der SSc sind die für die Grunderkrankung typische PAH, kardiale Insuffizienz, pulmonale Infekte, gastroösophagealer Reflux, Kachexie undAnämie.
Screening
Auf eine interstitielle Lungenerkrankung soll bei SSc-Patienten schon bei der Erstdiagnose gescreent werden. Diesbezüglich gibt es eindeutige Empfehlungen aus einem europäischen Consensus-Statement von 2020. Neben Klinik und Auskultation muss bei der Erstdiagnose in jedem Fall eine Lungenfunktion inkl. FVC und DLCO sowie eine HR-CT der Lunge durchgeführt werden. Auch in den Verlaufskontrollen soll regelmäßig an einen Lungenbefall gedacht werden, insofern ist eine regelmäßige Lungenfunktionsmessung empfohlen, das Intervall richtet sich nach der Einschätzung des Risikos für die Entwicklung bzw. Progression einer ILD. Die Frequenz von wiederholten HR-CTs richtet sich nach dem Schweregrad und der Progression der Lungenbeteiligung. Einer befürchteten hohen Strahlenbelastung durch die CT kann der Einsatz von Low-Dose-CT-Untersuchungen entgegengehalten werden. Der Einsatz von Biomarkern sowie einer Lungensonografie ist in der Praxis noch Gegenstand von Diskussionen.
Ösophagusbeteiligung
Etwa 60–90% der SSc-Patienten weisen einen gastroösophagealen Reflux auf, der in ca. 50% der Fälle symptomlos verläuft. Zu einem hohen Prozentsatz ist eine Ösophagusbeteiligung auch mit der Entwicklung bzw. Verschlechterung einer ILD assoziiert. Als Gründe hierfür werden einerseits die möglicherweise gleichzeitig vorliegende Beteiligung von Ösophagus und Lunge, andererseits auch Mikroaspirationen angenommen, die zu einer stetigen, chronischen Schädigung des Lungenepithels führen, was in letzter Konsequenz zu einer Lungenfibrose führt. Ebenso wurde postuliert, ob der erhöhte Ösophagusdiameter, der mit der Lungenfibrose einhergeht, Folge von intrathorakalen Ziehkräften ist, die somit zu einer Ösophagusdilatation führen. Ob der Einsatz von z.B. Protonenpumpeninhibitoren (PPI) zur Prävention/Therapie einer SSc-ILD Erfolgversprechend ist, wurde leider noch nicht untersucht.
Radiologische Bilder
Das häufigste radiologische Bild einer SSc-ILD ist mit ca. 78% die NSIP („non-specificinterstitialpneumonia“) (Abb. 3). Hier unterscheidet man histologisch einen entzündlichen Typ von einem entzündlich-fibrotischen Typ. Die UIP („usualinterstitialpneumonia“) kommt seltener vor und ist histologisch v.a. durch fibrotische Veränderungen gekennzeichnet. Je mehr Fibrose vorliegt, umso schlechter ist die Langzeitprognose.
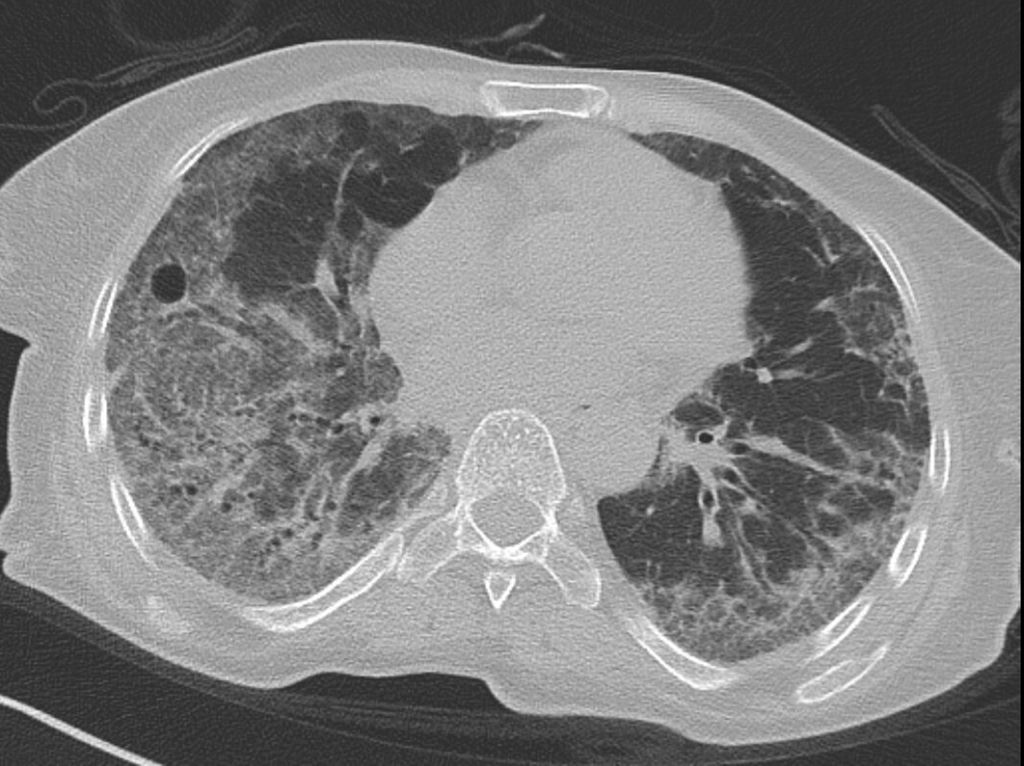
Abb. 3: HR-CT-Bild einer SSc-ILD („non-specific interstitial pneumonia“; NISP)
Therapie
Allgemeine Maßnahmen in der Behandlung der SSc-ILD sollten nie vergessen werden. Physiotherapie, Infektionsprophylaxe (Impfungen) und evtl. Rehabilitationsaufenthalte müssen dem Patienten regelmäßig angeboten werden.
Bezüglich einer etwaigen medikamentösen Behandlung sind im Vorhinein einige Fragen zu klären.
Wer wird womit therapiert?
Zuerst muss erhoben werden, ob ein zusätzlicher Organbefall vorliegt, der einer Immunsuppression bedarf, um mitunter beide oder mehrere Organe gleichzeitig behandeln zu können. Weiters muss mit den Patienten besprochen werden, welcher Behandlungserfolg (Symptomverbesserung oder nur -stabilisierung) und welche Nebenwirkungen zu erwarten sind. Essenziell ist auch das Erheben von Risikofaktoren für eine rasche Progression der ILD (Tab. 1).

Tab. 1: Risikofaktoren für eine rasche Progression der ILD
Anhand des Ausmaßes sowie der Progredienz im zeitlichen Verlauf ergeben sich die Notwendigkeit bzw. die Wahl der Therapie bzw. therapeutischen Möglichkeiten. So soll – grob vereinfacht – in Phasen eines hauptsächlich entzündlichen Bildes an eine antiinflammatorische bzw. immunsuppressive Therapie gedacht werden. Bei v.a. fibrotischem Bild werden zunehmend antifibrotische Medikamente verwendet.
Ab wann wird therapiert?
Die derzeitigen Empfehlungen sehen eine medikamentöse Therapie erst vor, wenn eine progresssive ILD vorliegt. Aufgrund des sehr heterogenen Verlaufs und der schlechten Vorhersagbarkeit der SSc-ILD ist dadurch einerseits ein regelmäßiges Follow-up notwendig, andererseits sollte auch die Risikoeinschätzung für einen schweren Verlauf mit einfließen. Ob diese Praxis in Zukunft so haltbar sein wird, ist fraglich, v.a. da nachgewiesen ist, dass es bei circa der Hälfte der Patienten in den ersten drei Jahren nach Diagnosestellung zu substanziellen Verschlechterungen der Lungenfunktion kommt.
Immunsuppressive Therapie
Die Grundlage für die Verwendung von Cyclophosphamid oder Mycophenolat-Mofetil (MMF) wurde in den Scleroderma-Lung-Studies I und II (SLS-I und -II) geschaffen. Diese beiden Medikamente haben weiterhin in der modernen Therapiestrategie ihren Platz. Rezent wurde in den USA der IL-6-Rezeptorblocker Tocilizumab für die Behandlung der SSc-ILD zugelassen. Für Rituximab gibt es bis dato noch sehr wenig Evidenz, obwohl dieses Medikament in der Praxis sehr oft (auch sehr oft erfolgreich) angewendet wird.
Antifibrotische Therapie
Mit der Veröffentlichung der SENSCIS-Studie im Jahr 2019 wurde durch den Beweis der Wirksamkeit von Nintedanib ein zusätzlicher Therapiearm erschlossen. V.a. in Kombination mit immunmodulatorischen Medikamenten (allem voran MMF) zeigte sich eine gute Wirksamkeit von Nintedanib im Sinne einer Verlangsamung der Verschlechterung der Lungenfunktion.
Ultima Ratio
Bei schweren Verlaufsformen und ausgesuchten Patienten kann weiterhin eine autologe Stammzelltransplantation durchgeführt werden, hier ist allerdings die therapiebedingte Mortalität mit 5–10% sehr hoch. Ebenso kann als Ultima Ratio eine Lungentransplantation erwogen werden.
Zusammenfassung
Insgesamt kann man feststellen, dass es in den letzten Jahren auf jeden Fall zu signifikanten Fortschritten in der Behandlung von Patienten mit einer SSc-ILD gekommen ist. Allerdings sind noch viele offene Fragen zu klären.
Literatur:
beim Verfasser
Das könnte Sie auch interessieren:


Das Pollenjahr 2025
Die Pollen fliegen wieder – und Allergiker:innen spüren das zurzeit massiv. Wertvolle Informationen zu Pollenflug, Pollenallergien und auch dem Einfluss von Luftschadstoffen auf ...

Gewebeschädigung: Proteasen bahnen der Allergie den Weg
Warum entwickeln manche Menschen Allergien und andere nicht? Viele Aspekte dieser Frage sind nach wie vor ungeklärt. Auf der klinischen Seite zeigt sich zunehmend, dass die Behandlung ...