.jpg)
20 Jahre idiopathische pulmonale Fibrose
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die idiopathische pulmonale Fibrose (IPF) ist der Hauptvertreter der idiopathischen interstitiellen Pneumonien (IIP). 2002 haben die amerikanische und die europäische Lungengesellschaft erstmals eine Klassifikation der interstitiellen Lungenerkrankungen erarbeitet, die – geringfügig modifiziert – im Prinzip immer noch gültig ist.
Vor 20 Jahren, also 2002, haben die amerikanische (ATS) und die europäische (ERS) Lungengesellschaft erstmals eine Klassifikation der interstitiellen Lungenerkrankungen erarbeitet und publiziert.1 In dieser Publikation wird auf die Wichtigkeit der Abgrenzung der idiopathischen pulmonalen Fibrose (IPF) von den anderen idiopathischen interstitiellen Pneumonien (IIP) hingewiesen, da die IPF von allen IIP die schlechteste Prognose hat (medianes Überleben <3 Jahren).
Definition
Die IPF ist definiert als eine bestimmte Form einer „chronisch progredienten fibrosierenden interstitiellen Pneumonie“, die primär bei älteren Patienten auftritt, auf die Lunge beschränkt ist und mit dem histopathologischen und/oder radiologischen Muster einer UIP („usual interstitial pneumonia“) einhergeht.2
Die IPF ist immer eine Ausschlussdiagnose. Daher haben die amerikanische, die europäische, die japanische und die südamerikanische Lungengesellschaft (ATS/ERS/JRS/ALAT) 2018 in einer „Clinical Practice Guideline“ zwei „motherhood statements“ aufgestellt:3
Alle Patienten mit Verdacht auf IPF müssen auf Umweltnoxen und Connective Tissue Diseases gescreent werden, da sich auch andere interstitielle Lungenerkrankungen mit einem UIP-Muster präsentieren können.
Die Diagnostik der IPF stellt immer noch eine große Herausforderung für Kliniker, Radiologen und Pathologen dar und kann nur in einem multidisziplinären Zugang erfolgen.
Daher werden seit über zehn Jahren an allen Lungenabteilungen Österreichs sogenannte „ILD(Interstitial Lung Disease)-Boards“ etabliert, um gemeinsam zu einer möglichst exakten klinischen Diagnose zu kommen. Grundlage für diese ILD-Boards war die Publikation von Flaherty 2004, der zeigen konnte, dass die interdisziplinäre Zusammenarbeit bzw. ein Informationsaustausch zielführend ist in Hinblick auf eine passende Diagnose.4
.jpg)
Abb. 1: CT-Bild einer UIP mit einer interstitiellen Verdickung, Verzerrung der Architektur, Wabenbildung und Bronchiektasie („CT scan in usual interstitial pneumonia [UIP]“ by Yale Rosen is licensed under CC BY 2.0; https://www.flickr.com/photos/pulmonary_pathology/4744513424/ )
Wann sollen wir den Verdacht auf eine IPF stellen?
Ein typischer IPF-Patient („appropriate clinical setting“) ist männlich, über 60 Jahrealt, Raucher oder Exraucher,klagt über Reizhusten und langsam zunehmende Belastungsdyspnoe und präsentiert sich klinisch auskultatorisch mit einem Knisterrasseln („velcro rales“), vielleicht auch mit Uhrglasnägeln und Trommelschlegelfingern.
HRCT als Goldstandard
Galt noch vor 20 Jahren die chirurgische Lungenbiopsie als „Goldstandard“ in der Diagnostik der IPF, so hat sich in den letzten 10–12 Jahren das Gewicht zugunsten der Radiologie verlagert: Die hochauflösende Computertomografie (HRCT) des Thorax (oder Dünnschicht-CT) ist in das Zentrum der Diagnostik gerückt.
So kann laut einem 2011 publizierten internationalenDiagnose-Algorithmus bei Vorliegen eines typischen/definitiven UIP-Musters in der HRCT auf eine invasive bioptische Diagnostik verzichtet werden.2
In der HRCT werden vier Muster unterschieden:3
-
definitives UIP-Muster,
-
wahrscheinliches UIP-Muster,
-
unbestimmt für UIP-Muster und
-
alternatives Muster (unvereinbar mit UIP).
Ein typisches UIP-Muster ist gekennzeichnet durch eine basale und subpleural betonte Prädominanz, retikuläre Verschattungen, Traktionsbronchiektasien und Honigwaben (Abb. 1). In Abhängigkeit vom vorliegenden Muster werden dann weitere diagnostische Schritte eingeleitet.
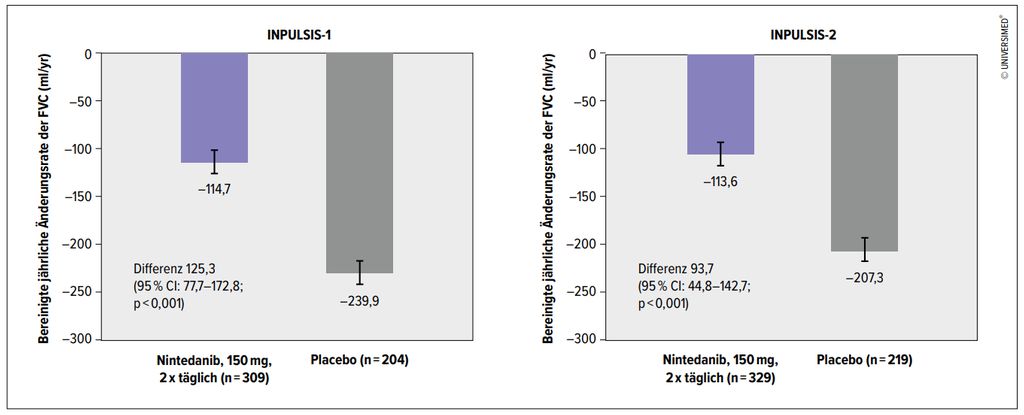
Abb. 2: Nintedanib vs. Placebo: Ergebnisse der Studien INPULSIS-1 und -2 (modifiziert nach Richeldi L et al. 2014)14
Invasive Diagnostik
Nur bei Vorliegen eines wahrscheinlichen oder unbestimmten UIP-Musters oder einer anderen alternativen radiologisch nicht klassifizierbaren Fibrose wird eine invasive Diagnostik empfohlen. Als Kliniker weise ich darauf hin, dass in der klinischen Praxis nur ca. 15% der infrage kommenden Patienten einer chirurgischen Lungenbiopsie zugeführt werden können. Als Kontraindikationen gelten:
-
eine Ruhehypoxämie spO2 <90% mit Raumluft,
-
DLCO <40% vom Soll,
-
TLC <50% vom Soll,
-
pulmonale Hypertension (sPAP >40mmHg) und
-
schwere Komorbiditäten.
Wir müssen auch immer bedenken, dass wir den Patienten durch eine chirurgische Lungenbiopsie auch der Gefahr einer akuten Exazerbation der Lungenfibrose aussetzen!
Wenn wir Pneumologen von einer invasiven bronchologischen Diagnostik bei Verdacht auf IPF sprechen, so meinen wir die Entnahme einer bronchoalveolären Lavage (BAL), einer transbronchialen Lungenbiopsie odereiner transbronchialen Kryobiopsie.
Bronchoalveoläre Lavage (BAL)
Grundsätzlich ist die Entnahme einer BAL fast allen Patienten zumutbar; es muss aber festgehalten werden, dass es kein typisches BAL-Muster für IPF gibt. Meist finden sich eine Erhöhung der Neutrophilen (ca. 10%) und eine leichte Eosinophilie (ca. 7,5%):Bei Vorliegen einer Lymphozytose (>20%) sollte jedenfalls an andere ILD gedacht werden (z.B. Sarkoidose, exogen-allergische Alveolitis, NSIP, Asbestose…). In diesem Setting dient die BAL zur Differenzialdiagnose und zum Ausschluss von Infektionen.
Im internationalen Diagnose-Algorithmus von 2011 wird die BAL gar nicht erwähnt, erst 2018 wird sie empfohlen, wenn kein typisches UIP-Muster in der HRCT vorliegt. In der deutsch-österreichischen S2k-Leitlinie zur Diagnostik der IPF hat man jedenfalls eine Empfehlung für die BAL bei IPF-Verdacht ausgesprochen.5
Transbronchiale Lungenbiopsie (TBLB)
Die transbronchiale Lungenbiopsie (TBLB) hat in der Regel eine sehr geringe diagnostische Ausbeute bei Verdacht auf IPF und wird daher in keiner internationalen oder nationalen Leitlinie empfohlen.
Transbronchiale Kryobiopsie (TBLC)
Seit vielen Jahren steht die transbronchiale Kryobiopsie (TBLC) im Fokus der Bronchologie, stellt sie doch eine mögliche Alternative zur chirurgischen Lungenbiopsie bei Verdacht auf IPF dar. Dabei wird das peripher und subpleural gelegene Lungenparenchym im Rahmen einer Bronchoskopie mit einer Kryosonde „angeeist“: Es können dadurch größere Bioptate gewonnen werden, die auch keine Quetschartefakte aufweisen und daher vom Pathologen besser befundet werden können.
Die internationale „Clinical Practice Guideline“ 2018 hat diese Methode noch nicht empfohlen, in einem Update 2022 aber aufgrund neuer Studien (z.B. COLDICE6) in den Diagnose-Algorithmus bei Verdacht auf IPF vor einer chirurgischen Lungenbiopsie aufgenommen.
Folgende Sicherheitsaspekte sind bei der Entnahme einer TBLCzu beachten: Die TBLC hat ein erhöhtes Risiko für eine postbioptische Blutung und Pneumothorax und sollte daher nur in erfahrenen Bronchologie-Zentren unter Einhaltung bestimmter Sicherheitsstandards durchgeführt werden.7
Von der Diagnose zur Therapie
Vor 20 Jahren war die IPF eine fatale Erkrankung mit sehr schlechter Prognose (medianes Überleben <3 Jahren), z.T. schlechter als bei vielen Malignomen (nur Lungen- und Pankreaskarzinom hatten eine schlechtere Prognose).
Ein Joint Statement von ATS/ERS vom Jahr 2000 empfiehl – auf der Basis von Expertenmeinungen – eine kombinierte Therapie mit oralen Steroiden und Azathioprin (alternativ Cyclophosphamid).8
Grundsätzlich hatten die Therapiekonzepte von 2000 bis 2012 einen antiinflammatorischen/immunsuppressiven Ansatz, ab 2005 ergänzt durch eine antioxidative Therapie mit hochdosiertem N-Acetylcystein9 – dies war die sogenannte Tripeltherapie, die ab 2005 auch in Österreich breit angewendet wurde.
Als Kliniker habe ich damals schon den Eindruck gehabt, dass diese Therapie keine positiven Effekte, aber viele Nebenwirkungen gezeigt hat. Dieser Eindruck wurde wissenschaftlich durch die PANTHER-Studie bestätigt, die ein erhöhtes Risiko für Tod und Hospitalisierung für Patienten unter Tripeltherapie zeigte.10 Seitdem gilt die Tripeltherapie als obsolet in der Behandlung der IPF.
Daneben gab es Studien mit antifibrotisch wirkenden Substanzen wie Interferon gamma, Colchicin, TNF-alpha-Antagonisten, Endothelinrezeptorantagonisten, TGF-beta-1-Antagonisten und auch Pirfenidon. Alle diese Studien zeigten zwar einen positiven Trend an, erzielten aber letztlich keinen erforderlichen Evidenzgrad.
Pirfenidon und Nintedanib
Ein Durchbruch in der Therapie der IPF war die Publikation von Taniguchiim Jahr 2010, der in einer Phase-III-Studie die Wirksamkeit von Pirfenidon zeigen konnte, gemessen am verzögerten FVC-Abfall vs. Placebo.11 Persönlich würde ich diese Studie als „Silberstreif am Horizont“ für die Therapie der IPF ansehen. Aus formalen Gründen wurde die Taniguchi-Studie von der FDA in den USA nicht anerkannt, es wurden weitere Pirfenidon-Studien für die Zulassung gefordert: Die CAPACITY-Trials bestätigten die Wirksamkeit von Pirfenidon; primärer Endpunkt war – wie in allen weiteren Zulassungsstudien – der verzögerte Abfall der FVC vs. Placebo.12
Parallel zu Pirfenidon hat sich eine andere Substanz in der wissenschaftlichen Forschung herauskristallisiert: Nintedanib, ein Tripeltyrosinkinaseinhibitor, der die Fibroblastenproliferation hemmt und somit die zunehmende Fibrosierung des Lungenparenchyms positiv beeinflusst. In der Phase-II-TOMORROW-Studie konnte durch Nintedanib eine Reduktion der Exazerbationen der IPF und des FVC-Abfalls gezeigt werden.13 Diese Studie war die Basis für die nachfolgenden Phase-III-Studien INPULSIS-1 und -2 (Abb. 2).14 Zeitgleich istdie ASCEND-Studie publiziert worden, die die Wirksamkeit von Pirfenidon bestätigte.15 Die Ergebnisse der INPULSIS- und ASCEND-Studien haben 2014 zur Zulassung von Pirfenidon bzw. Nintedanib für die Therapie der IPF durch die FDA in den USA geführt. In Österreich gibt es eine Zulassung für Pirfenidon seit 2012, für Nintedanib seit 2015. Die Kosten für diese oral verfügbare Therapie werden von allen Kassen übernommen.
Wir sind als Pneumologen in der glücklichen Lage, dass wir durch zwei zugelassene antifibrotische Medikamente die Prognose unserer IPF-Patienten deutlich verbessern können. Viele meiner Patienten sind schon seit vielen Jahren unter diesen Therapien.
Fazit
Ich meine, dass es in den letzten 20 Jahren durch wissenschaftliche Fortschritte gelungen ist, die IPF von einer „fatalen“ Lungenerkrankung zu einer „chronischen“ Lungenerkrankung zu machen. Der Beitrag der Radiologie (HRCT-Thorax) ist zentral in der Diagnostik, gemeinsam mit der transbronchialen Kryobiopsie wurde die chirurgische Lungenbiopsie im Diagnose-Algorithmus verdrängt. Die multidisziplinäre Diagnose (ILD-Board) hat sich als Goldstandard in der klinischen Praxis bewährt.
Literatur:
1 ATS/ERS: Am J Respir Crit Care Med 2002; 165(2): 277-304 2 Raghu G et al.: Am J Respir Crit Care Med 2011; 183(6): 788-824 3 Raghu G et al.: Am J Respir Crit Care Med 2018; 198(5): e44-e68 4 Flaherty KR et al.: Am J Respir Crit Care Med 2004; 170(8): 904-10 5 Behr J et al.: S2k-Leitlinie 2017; AWMF-Register Nr. 020/025 6 Troy LK et al.: Lancet Respir Med 2020; 8(2): 171-81 7 Hetzel J et al.: Respiration 2018; 95(3): 188-200 8 ATS/ERS: Am J Respir Crit Care Med 2000; 161(2 Pt 1): 646-64 9 Demedts M et al.: N Engl J Med 2005; 353: 2229-42 10 The Idiopathic Pulmonary Fibrosis Clinical Research Network: N Engl J Med 2012; 366: 1968-77 11 Taniguchi H et al.: Eur Respir J 2010; 35: 821-9 12 Noble PW et al.: Lancet 2011; 377(9779): 1760-9 13 Richeldi L et al.: N Engl J Med 2011; 365: 1079-87 14 Richeldi L et al.: N Engl J Med 2014; 370: 2071-82 15 King TE et al.: N Engl J Med 2014; 370: 2083-92
Weitere Literatur:
beim Verfasser
Das könnte Sie auch interessieren:

Best of DGP: Kongress-Highlights für die Praxis
Dr. Sabine Lampert, niedergelassene Pneumologin in Erlangen, fasste die Erkenntnisse vom DGP-Kongress 2025 für die tägliche Praxis zusammen. Sie konzentrierte sich bei ihrem „Best of DGP ...
Lungenembolie: Engramme für den Behandlungspfad
Die Lungenembolie ist ein häufiges und potenziell lebensbedrohliches Krankheitsbild. Die Diagnose bleibt herausfordernd – immer noch zählt die Lungenembolie zu den Diagnosen, die am ...
