
20 Jahre zystische Fibrose
Seit mehr als 70 Jahren ist die zystische Fibrose (CF) als eigenständiges Krankheitsbild bekannt. Damals noch mit einer äußerst schlechten Überlebensprognose assoziiert, kann die CF heute mit einem interdisziplinären Behandlungsansatz gut kontrolliert werden. In den letzten 20 Jahren konnte dadurch die Lebenserwartung der CF-Patienten enorm gesteigert werden.
Es war ein warmer Junitag Ende der 1970er-Jahre. Zum Schwitzen brachte mich jedoch nicht der Frühsommer, sondern das bevorstehende Rigorosum für Kinderheilkunde. Wie üblich kam nach den Fragen der Patient. Mein Patient war ein 5-jähriger Bub – viel zu klein, viel zu dünn mit viel zu großem Bauch. Immer wieder wurde der Kleine von Hustenanfällen geschüttelt und seine Mutter forderte ihn auf, den Schleim – dunkelgrün und zäh – auszuspucken. Nach kurzem Gespräch mit der Mutter gab es keinen Zweifel an der Diagnose: zystische Fibrose (CF). Der Patient wurde wieder aus dem Hörsaal entlassen. Mein Prüfer bestätigte die Diagnose und meinte, dass der Bub wahrscheinlich noch seine Einschulung erleben wird, sicher jedoch seine Volksschulzeit nicht überleben wird. Beklemmende Stille erfüllte den Hörsaal. So sehr ich mich auch über mein „Bestanden“ freute, so klar war auch: Kinderärztin werde ich nie. Nein, so ohnmächtig und hilflos schwerstkranken Kindern gegenüberzustehen, das möchte ich nicht. So dachte ich, nicht ahnend, dass CF mein berufliches Lebensthema werden sollte.
Dankbar und demütig freue ich mich heute, dass durch den enormen Anstieg der Lebenserwartung das erste CF-Erwachsenenzentrum in Österreich an unserer Abteilung etabliert werden konnte. Ebenso freue ich mich, die gewaltigen Fortschritte in der Betreuung und Therapie der CF-Patienten miterlebt zu haben. Zuerst jedoch ein Blick zurück…
20. Jahrhundert
Bereits Anfang der 1950er-Jahre kannte man CF als eigenständiges Krankheitsbild. Die Prognose war fatal, nur jedes zweite Kind erlebte seinen zweiten Geburtstag. Mitte der 1950er-Jahre entstanden die ersten Zentren und es wurden intensive Behandlungskonzepte entwickelt. Ernährung, Atemwegsreinigung und aggressive Infektionsbehandlung wurden als Grundsäulen der Therapie erkannt. Daran hat sich bis heute nichts geändert, jedoch hat sich jede Säule enorm weiterentwickelt.
Ernährung
Fettfreie Ernährung stand am Anfang, da durch die Pankreasinsuffizienz keine Fettverdauung möglich ist. So war bei erhöhtem Energiebedarf durch chronische Entzündung und Infektion keine adäquate Ernährung möglich. Mangelernährung und Kachexie waren die Folgen, die den unseeligen Kreislauf weiter antrieben. Mit den ersten Pankreasenzymen gelang der Durchbruch, und man konnte den Ernährungszustand der Kinder massiv verbessern. Dies führte auch zu einem ersten steilen Anstieg der Lebenserwartung (Abb.1).
Heute haben wir mikroverkapselte Pankreasenzyme zur Verfügung, somit ist die Therapie dem Fettgehalt entsprechend sehr gut zu dosieren, und die Elastasemessung im Stuhl ermöglicht ein gutes Monitoring. Während früher das Motto hieß: „Viel essen, egal was, je fetter, umso besser“, ist auch bei CF die gesunde Ernährung angekommen. Heute wird eine protein- und ballaststoffreiche Nahrung mit hochwertigen Fetten empfohlen. Mangelernährung, Kachexie gehören der Vergangenheit an. Nahezu alle Patienten können heute einen normalen Ernährungszustand erreichen.
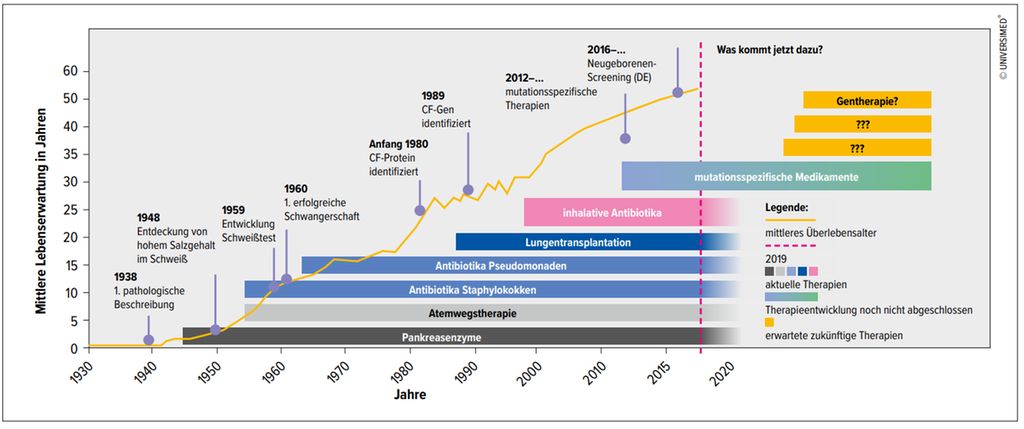
Abb. 1: Entwicklung der Lebenserwartung von CF-Betroffenen von 1930 bis heute (Quelle: Deutsches Mukoviszidose-Register, Patientenberichtsband 2020)
Atemwegsreinigung
Zur Reinigung der Atemwege dienen die Inhalationstherapie, die Physiotherapie und Sport.
Inhalationstherapie
Standen anfangs nur 0,9%iges NaCl zur Befeuchtung und Beta-2-Mimetika zur Verfügung, kamen in den 1990ern sehr wirksame Medikamente zur Verflüssigung des Schleimes und damit zur leichteren Expektoration auf den Markt. DNAse sowie hypertone Kochsalzlösungen haben heute einen fixen Platz in der Therapie.
Physiotherapie
Klopfen war der erste physiotherapeutische Versuch, das Sekret zu mobilisieren. Inzwischen eine obsolete Methode. Autogene Drainage, PEP („positive expiratory pressure“) und Kombinationen aus beiden sind heute die gängigsten Methoden. Physiotherapeuten lehren den Patienten die für sie passende Methode, das Sekret selbstständig zu mobilisieren. Bei akuten Infektexazerbationen und weit fortgeschrittenem Krankheitsbild ist die professionelle Unterstützung der Physiotherapeuten notwendig und hilfreich.
Sport
Dass sich Bewegung günstig auf den Krankheitsverlauf auswirkt, wusste man früh und empfahl Bewegung, „so wie es halt mit der Luft geht“. Heute können wir durch Spiroergometrie genau zwischen pulmonaler Limitierung und schlechtem Trainingszustand unterscheiden. Dies ermöglicht uns, individuelle Trainingspläne zu erstellen. Die Patienten sind oft überrascht, dass sie überhaupt nicht pulmonal eingeschränkt sind, sondern die Atemnot bei sportlicher Belastung schlicht durch einen schlechten Trainingszustand bedingt ist. Training ist, natürlich angepasst, auch im fortgeschrittenen Krankheitsstadium möglich und notwendig. Generell gilt – wie für uns alle – eine Kombination aus Ausdauer- und Krafttraining als ideal.
Antibiotikatherapie
Aggressive Antibiotikatherapie war immer schon ein ganz wesentlicher Therapiebestandteil. Während Staphylokokken gut oral zu behandeln waren und sind, sieht die Situation beiPseudomonas anders aus. Fast alle Medikamente sind i.v. zu verabreichen. Bei Patienten mit weit fortgeschrittener Erkrankung hat man die Erfahrung gemacht, dass die i.v.Antibiotika auch inhalativ sehr gut wirken. Allerdings enthalten diese Medikamente potenziell inhalativ toxische Lösungsmittel, sodass ein breiter Einsatz nicht möglich war.
Anfang dieses Jahrhunderts kamen dann die ersten inhalativen Antibiotika auf den Markt, sicherlich ein Meilenstein in der Pseudomonastherapie. Sowohl aus der Behandlung von akuten Infektionen als auch als Dauertherapie im Sinne einer Suppressionstherapie sind die inhalativen Antibiotika nicht mehr wegzudenken.
Transplantationsmedizin: LuTx
Anfang der 90er wurden in Wien die ersten Lungentransplantationen (LuTx) durchgeführt mit noch eher bescheidenen Erfolgen. Bei den Aufklärungsgesprächen sprach ich damals in erster Linie von der Möglichkeit der Verbesserung der Lebensqualität durch eine LuTx und eventuell auch einer Lebensverlängerung. Die Patienten sahen diese Therapieoption eher skeptisch und viele lehnten das Therapieangebot ab. Ganz anders die Situation heute. Die 5-Jahres-Überlebenszeit und darüber hinaus ist in den letzten 20 Jahren massiv angestiegen. Viele erreichen nach der Transplantation eine sensationelle Leistungsfähigkeit und Lebensqualität. Die Expedition auf den Kilimandscharo mit Lungentransplantierten 2017 ist eine beeindruckende Bestätigung der Erfolgsstory Transplantationsmedizin. Heute gibt es kaum Patienten, die die Option einer LuTx ablehnen.
Modulatoren
Ja, es hat sich viel getan, und die Lebenserwartung und die Lebensqualität kletterten ständig nach oben (Abb. 1). Jedoch waren ausschließlich symptomatische Therapien möglich, ein ständiges Hinterherlaufen. Wann wird es endlich eine kausale Therapie geben? Diese Frage wurde oft von den Patienten gestellt. Mit Entdeckung des CF-Gens 1989, so glaubte man, ist es zur Gentherapie nicht mehr weit. Ein Irrtum, wie sich zeigte. Die Forschungen am CFTR(„Cystic fibrosis transmembrane conductance regulator“)-Kanal und die Entwicklung funktionsverbessernder Medikamente zeigten hingegen Erfolg.
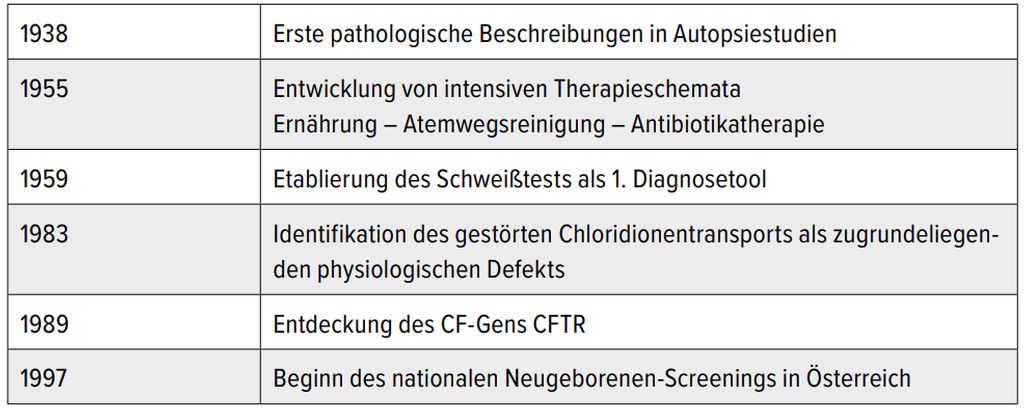
Tab. 1: Historischer Überblick
2012 war es so weit, die erste kausale Therapie, der erste Modulator, Ivacaftor, war in Österreich erhältlich. Inzwischen stehen vier Präparate zur Auswahl (Tab.2). Die Verordnung erfolgt der individuellen Genetikentsprechend. Für etwa 85% der CF-Patienten steht derzeit ein Modulator zur Verfügung. Prinzipiell ist zu beachten, dass die Modulatoren eine zusätzliche Therapie sind und nicht die symptomatische Therapie ersetzen. Sämtliche Studien mit ihren guten, teilweise sensationellen Ergebnissen wurden unter Beibehaltung der Therapie durchgeführt. Die Funktion des CFTR-Kanals kann maximal um 50% verbessert werden. Bereits bestehende Strukturschäden bleiben irreparabel. Auch neue Keimbesiedelungen können durch die Modulatoren nicht verhindert werden. Apropos sensationelle Ergebnisse: Je nach Modulator zeigen sich FEV1-Anstiege um 4–14%, Verringerungen der Exazerbationen um 30–63% sowie deutliche Gewichtszunahmen und eine signifikante Verbesserung der Lebensqualität.
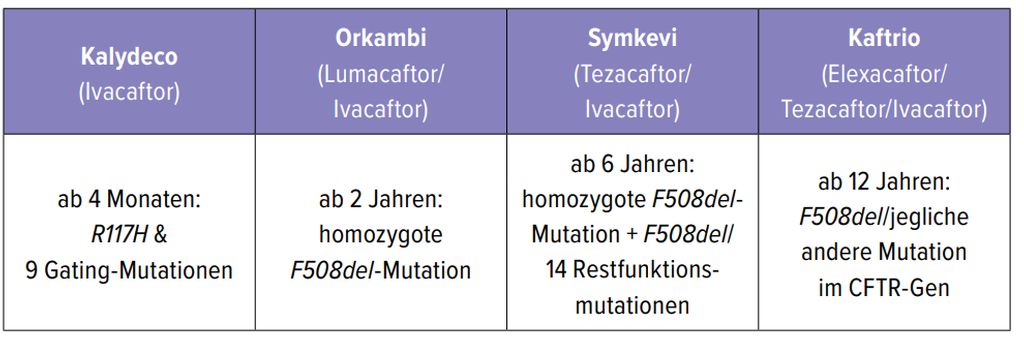
Tab. 2: Überblick über die zur Verfügung stehenden Modulatoren
Fazit
Die Modulatorentherapie als erste kausale Therapie ist sicherlich das Highlight der Therapie der letzten 20 Jahre. Ziel ist es, in Kürze allen CF-Patienten einen Modulator anbieten zu können. Die bisherige symptomatische Therapie muss beibehalten werden. Ebenso ist weiterhin eine Droge unverzichtbar: die „Droge“ Arzt bzw. die „Droge“ CF-Team. Diese Drogen sind hochselbstwirksam und wirken als Katalysatoren aller anderen Therapien. Nur durch intensive empathische Begleitung, Unterstützung und Motivation können die Patienten von den Errungenschaften der Forschung profitieren. Für die Zukunft wünsche ich mir, dass CF wieder ein ausschließlich pädiatrisches Thema wird. Ich wünsche mir, dass CF heilbar wird, und zwar im frühen Kindesalter. Für alle anderen Ärzte soll CF nur mehr eine anamnestische Notiz sein. Mein Traum ist, einen Vortrag zu hören oder einen Artikel zu lesen mit dem Titel: „Wie CF heilbar wurde“. Eine Utopie? Vielleicht. Aber jeder Fortschritt, jede Errungenschaft hat mit einer Utopie begonnen.
Literatur:
bei der Verfasserin
Das könnte Sie auch interessieren:

COPD: neue Leitlinie für eine bessere Patientenversorgung
Die aktualisierte S2k-Leitlinie „Fachärztliche Diagnostik und Therapie der chronisch obstruktiven Lungenerkrankung (COPD) 2026“ wurde im Februar publiziert und beim DGP-Kongress im März ...

Mit welchen Pollen man nun rechnen muss
Kälteeinbruch und Regen hatten die Pollenbelastung stark gebremst. Mit Ostern stiegen nun die Temperaturen stark und damit steigen nun die Pollenbelastungen durch Esche, Forsythie und Birke.