
„Diagnostic delay“ und Überleben bei Alpha-1-Antitrypsin-Mangel
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Alpha-1-Antitrypsin-Mangel (AATM) ist eine unterdiagnostizierte Krankheit und die verzögerte Diagnosestellung ist ein häufiges Problem. Dass dies zu einem schlechteren Outcome führen könnte, wurde schon lange vermutet. Nun konnte bei einer Auswertung des österreichischen Registers erstmals gezeigt werden, dass eine verzögerte AATM-Diagnose mit einem kürzeren Gesamtüberleben und transplantationsfreien Überleben assoziiert ist. Die Arbeit wurde im wissenschaftlichen Journal „Respiratory Research“ publiziert.1
Keypoints
-
Alpha-1-Antitrypsin-Mangel (AATM) ist eine unterdiagnostizierte Krankheit und die verzögerte Diagnosestellung ist ein häufiges Problem.
-
In unserer Kohorte war eine verzögerte AATM-Diagnose mit schlechterem Gesamtüberleben und transplantationsfreiem Überleben assoziiert, unabhängig von Alter, Rauchverhalten, BMI, Lungenfunktion (FEV1) und der Notwendigkeit einer Langzeit-Sauerstoffgabe (LTOT).
-
Daher sollten – in Übereinstimmung mit den relevanten Guidelines – alle Patient*innen mit COPD, Emphysem, untypischem Asthma bronchiale und Verwandte von AATM-Patient*innen auf AATM getestet werden.
Hintergrund
Alpha-1-Antitrypsin-Mangel (AATM) ist eine genetische Erkrankung, die sich u.a. als Lungen- oder Lebererkrankung manifestieren kann.2 Sie wird durch Mutationen im SERPINA1-Gen verursacht, das für den „Proteinase-Inhibitor“ (Pi) Alpha-1-Antitrypsin (AAT) kodiert.2 Die meisten Fälle von schwerem AATM entstehen durch das homozygote Auftreten der Punktmutation Glu342Lys („Z-Allel“), also durch den Pi*ZZ-Genotyp, der sowohl zu Lungen- als auch zu Lebermanifestationen führen kann und typischerweise durch einen niedrigen AAT-Serumspiegel gekennzeichnet ist.2–4 Schätzungen zufolge tragen über 100000 Personen in Europa den Pi*ZZ-Genotyp.5,6
Eine noch wesentlich häufigere Variante ist das „S-Allel“ (Glu264Val), das in heterozygoter Kombination mit dem Z-Allel (Pi*SZ) das Risiko für Leberfibrose und – insbesondere bei Rauchern – für COPD/Emphysem erhöht.7–9 Aktuelle Daten deuten darauf hin, dass auch Personen mit der heterozygoten Kombination von Z-Allel und dem „M-Allel“ genannten Wildtyp (Pi*MZ) ein erhöhtes Risiko für COPD und Leberzirrhose haben könnten, insbesondere in Kombination mit anderen Risikofaktoren, wobei diese Assoziation schwächer zu sein scheint als bei Pi*SZ.10–14
AAT hat in der Lunge die Funktion, die schädliche Einwirkung von Proteinasen, insbesondere der neutrophilen Elastase, auf das Lungengewebe zu unterbinden.2,15 Dementsprechend sind typische Lungenmanifestationen des AATM Emphysem und COPD, ausgelöst durch die von der ungehemmten Proteinase-Aktivität verursachte chronische Entzündung.2,15 Die unzureichende Menge von (funktionsfähigem) AAT wird durch Nikotinabusus noch weiter vermindert.16 Daher entwickeln viele Pi*ZZ-Patient*innen, die rauchen, ein Emphysem bzw. eine COPD in ungewöhnlich jungem Alter und das Risiko für diese Erkrankungen ist bei Raucher*innen mit Pi*SZ oder Pi*MZ signifikant erhöht gegenüber der Normalbevölkerung.8,9,12
Bereits in der Vergangenheit konnte gezeigt werden, dass die Diagnose AATM in vielen Fällen sehr verzögert gestellt wird, oft viele Jahre nach dem Symptombeginn.17,18 Auch wenn bereits diskutiert wurde, dass eine frühzeitige AATM-Diagnose hilfreich sein könnte, da sie zeitnahe Tabakentwöhnung, Screening der Familienangehörigen und die Einleitung einer AAT-Augmentationstherapie ermöglicht19, gab es bisher kaum direkte Belege der Schädlichkeit einer verzögerten AATM-Diagnose.
In einer Publikation aus dem Jahr 2019 konnte gezeigt werden, dass eine verzögerteAATM-Diagnose mit einem schlechteren klinischen Status und mit einem fortgeschrittenen Stadium der Lungenerkrankung assoziiert war.20 Bisher war jedoch – soweit uns bekannt – keine Studie über den Einfluss einer verzögerten AATM-Diagnosestellung auf das Überleben veröffentlicht worden.
Zielsetzung & Methoden
Ziel unserer Auswertung des nationalen„Austrian Alpha-1 Lung“(AAL)-Registers war es, den Einfluss einer verzögerten AATM-Diagnosestellung auf Gesamtüberleben, transplantationsfreies Überleben und klinische Charakteristika zu untersuchen.
Das AAL-Register ist das nationale österreichische Register zu AATM, zu dem neun spezialisierte Zentren (in alphabetischer Reihenfolge: Klinik Ottakring, Wien, Klinikum Klagenfurt am Wörthersee, Klinikum Wels-Grieskirchen, Landeskrankenhaus Hohenems, Landeskrankenhaus Natters,LKH Graz IIEnzenbach,LKH Graz II West, Medizinische Universität Innsbruck, Paracelsus Medizinische Universität, Salzburg) beigetragen haben. Alle erwachsenen AATM-Patient*innen, die eine schriftliche Einverständniserklärung unterzeichnen, können in das Register eingeschlossen werden.
„Diagnostic delay“ (übersetzt etwa „Verzögerung in der Diagnosestellung“) wurde definiert als die Zeitspanne vom ersten Auftreten respiratorischer Symptome bis zur AATM-Diagnose. Die Daten wurden mittels deskriptiver Statistik, Chi-Quadrat-Test und uni- und multivariabler Überlebensanalysen (Kaplan-Meier-Kurven, Log-Rank-Test, Cox-Regression) ausgewertet. Um die relevante Population zu untersuchen, wurden alle Analysen auf symptomatische AATM-Patient*innen bezogen, bei denen Daten zu Überleben und „diagnostic delay“im Register verfügbar waren. Im Detail ist die Methodik in der Publikation beschrieben.1
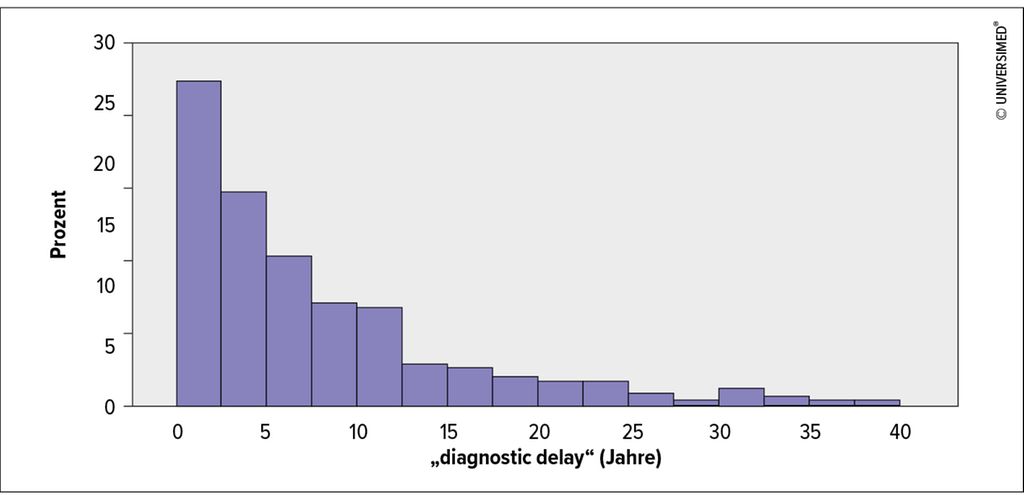
Abb. 1: Verteilung des „diagnostic delay“ (modifiziert nach Meischl T et al. 2023)1
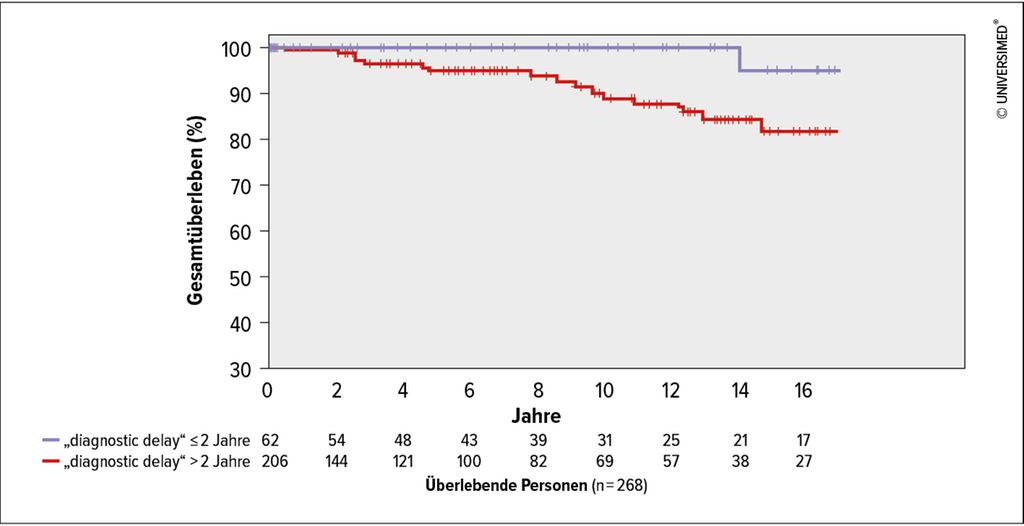
Abb. 2: Kaplan-Meier-Kurve des Gesamtüberlebens („overall survival“), stratifiziert nach „diagnostic delay“ ≤/>2 Jahre (modifiziert nach Meischl T et al. 2023)1
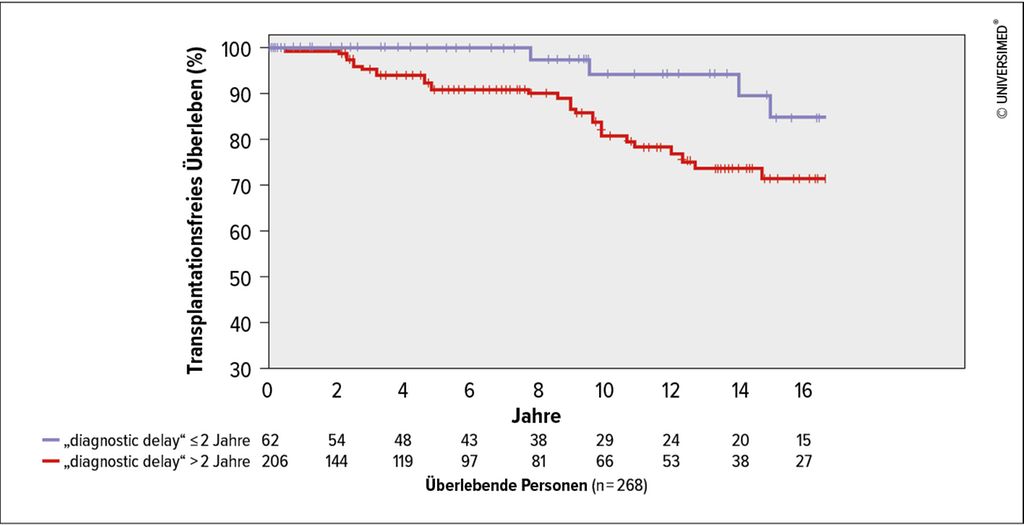
Abb. 3: Kaplan-Meier-Kurve des transplantationsfreien Überlebens („transplant-free survival“), stratifiziert nach „diagnostic delay“ ≤/>2 Jahre (modifiziert nach Meischl T et al. 2023)1
Ergebnisse
Der häufigste Genotyp in der ausgewerteten Kohorte war Pi*ZZ (82,1%). Bei Diagnose hatten 90,2% einen verminderten AAT-Spiegel (<0,6g/L). 28,2% der Patient*innen haben nie geraucht, 68,0% waren ehemalige Raucher*innen und 3,8% waren aktive Raucher*innen. Eine Lungenerkrankung war bei 98,5% diagnostiziert worden, in den meisten Fällen Emphysem (63,8%) und/oder COPD (44,0%). Der mediane „diagnostic delay“ lag bei 5,3 Jahren (Interquartilsabstand 2,2–11,5 Jahre). In der multivariablen Analyse war ein längerer „diagnostic delay“mit signifikant kürzerem Gesamtüberleben (Hazard-Ratio [HR]: 1,61; 95%-Konfidenzintervall: 1,09–2,38; p=0,016) und transplantationsfreiem Überleben (HR: 1,43; 95%-Konfidenzintervall: 1,08–1,89; p=0,011) assoziiert, unabhängig von Alter, Rauchverhalten, Body-Mass-Index (BMI), Lungenfunktion (FEV1) und Langzeit-Sauerstoffgabe (LTOT). Außerdem waren BMI, Alter und aktiver Nikotinabusus mit kürzerem Gesamtüberleben sowie BMI, aktiver Nikotinabusus und verminderte Lungenfunktion mit kürzerem transplantationsfreiem Überleben assoziiert. Weitere Ergebnisse sind in der Publikation angeführt.1
Diskussion & Conclusio
Bereits 2003 wurde in einem gemeinsamen Statement der American Thoracic Society (ATS) und der European Respiratory Society (ERS) empfohlen, alle Patient*innen mit Lungenemphysem, COPD, Asthma bronchiale mit inkomplett reversibler Obstruktion und unklarer Lebererkrankung sowie Verwandte von AATM-Patient*innen auf AATM zu testen21, ebenso wie in den Clinical Practice Guidelines der Alpha-1 Foundation22 und in den Empfehlungen der ERS aus dem Jahr 201723. Diese Empfehlungen scheinen jedoch nicht vollständig eingehalten zu werden, denn in einer britischen Kohorte von COPD-Patient*innen wurden nur 2% getestet, obwohl die AATM-Prävalenz unter den Getesteten 24% betrug.24 In einer anderen Studie wiesen 9% einer COPD-/Emphysem-Kohorte den Pi*ZZ-Genotyp auf.25 Insgesamt dürfte AATM eine unterdiagnostizierte Erkrankung sein und Screening-Programme auf Bevölkerungsebene würden Schätzungen zufolge eine*n asymptomatische*n Träger*in pro 3500 gescreenten Personen finden.5,6,26,27
Gründe dafür dürften unter anderem das mangelnde Wissen der Ärzt*innen über AATM und die Guidelines sowie die fehlende klinische Unterscheidbarkeit von „normaler“ COPD sein.26–28
Diese Hürden müssen überwunden werden: Zum einen ist aktiver Nikotinabusus – in unserer und in vielen anderen Studien – ein wichtiger Risikofaktor für ungünstiges Outcome bei AATM, und eine zeitgerechte Diagnose macht frühzeitige Tabakentwöhnung möglich, was die Entwicklung von Lungenerkrankungen verhindern oder zumindest verlangsamen kann.19,29 Eine schwedische Studie konnte zeigen, dass Personen mit Pi*ZZ-Genotyp eine geringere Lebenserwartung als die Normalbevölkerung hatten, die Pi*ZZ-Nichtraucher*innen jedoch eine fast idente Lebenserwartung wie die Normalbevölkerung aufwiesen; somit dürfte baldige Tabakentwöhnung eine der wichtigsten Maßnahmen nach AATM-Diagnosestellung sein.30 Zum anderen können Patient*innen, bei denen AATM frühzeitig diagnostiziert wird, von der (früheren) Einleitung einer AAT-Augmentationstherapie profitieren.31 Darüber hinaus können nach rascher AATM-Diagnose die Verwandten frühzeitig getestet werden, was bei diesen wiederum die Chance bietet, von einer frühzeitigen Lifestyle-Modifikation zu profitieren.27
Da ungezielte Screening-Verfahren wie z.B. Neugeborenen-Screening hohe Kosten bei geringer Rate identifizierter AATM-Träger*innen verursachen würden, werden vor allem gezielte Teststrategien bei Patient*innen mit Lungenerkrankungen diskutiert.19,24,27 Vorschläge, um die Detektionsrate bei AATM zu erhöhen, beinhalten unter anderem die flächendeckende Sequenzierung des SERPINA1-Gens bei COPD-Patient*innen, die Auswertung elektronischer Krankenakten mittels Big-Data-Analysen zur Identifizierung potenzieller AATM-Träger*innen und die Testung auf AATM direkt im Rahmen von Lungenfunktions-Untersuchungen.27,32,33
Zusammenfassend war in unserer Kohorte eine verzögerte AATM-Diagnosestellung mit kürzerem Gesamtüberleben und transplantationsfreiem Überleben assoziiert, unabhängig von Alter, Rauchverhalten, BMI, Lungenfunktion (FEV1) und der Notwendigkeit einer Langzeit-Sauerstoffgabe (LTOT).1 Daher sollten die Anstrengungen intensiviert werden, eine frühzeitige Diagnosestellung sicherzustellen. Insbesondere sollten – in Übereinstimmung mit den relevanten Guidelines – alle Patient*innen mit COPD, Emphysem und untypischem Asthma bronchiale sowie Verwandte von AATM-Patient*innen auf AATM getestet werden.
Literatur:
1Meischl T, Schmid-Scherzer K, Vafai-Tabrizi F et al.: The impact of diagnostic delay on survival in alpha-1-antitrypsin deficiency: results from the Austrian Alpha-1 Lung Registry. Respir Res 2023; 24(1): 34 2 Strnad P et al.: Alpha1-antitrypsin deficiency. N Engl J Med 2020; 382(15):1443-55 3 Jeppsson JO: Amino acid substitution Glu leads to Lys alpha1-antitrypsin PiZ. FEBS Lett 1976; 65(2):195-7 4 Hamesch K et al.: Liver fibrosis and metabolic alterations in adults with alpha-1-antitrypsin deficiency caused by the Pi*ZZmutation. Gastroenterology 2019; 57(3):705-19 5 Blanco I et al.: Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis 2017; 12:561-9 6 Greulich T et al.: The prevalence of diagnosed α1-antitrypsin deficiency and its comorbidities: results from a large population-based database. Eur Respir J 2017; 49(1): 1600154 7 Mandorfer M et al.: Liver disease in adults with α1-antitrypsin deficiency. United Eur Gastroenterol J 2018; 6(5):710-8 8 McElvaney G et al.: Clinical considerations in individuals with α1-antitrypsin PI*SZ genotype. Eur Respir J 2020; 55(6): 1902410 9 Franciosi AN et al.: Clarifying the risk of lung disease in SZ alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2020; 202(1): 73-82 10 Strnad P et al.: Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019; 68(6):1099-1107 11 Schaefer B et al.: Heterozygosity for the alpha-1-antitrypsin Z allele in cirrhosis is associated with more advanced disease. Liver Transplant 2018; 24(6):744-51 12 Molloy K et al.: Clarification of the risk of chronic obstructive pulmonary disease in α1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189(4):419-27 13 Foreman MG et al.: Alpha-1 antitrypsin PiMZ genotype is associated with chronic obstructive pulmonary disease in two racial groups. Ann Am Thorac Soc 2017; 14(8):1280-7 14 Ferkingstad E et al.: Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat Commun 2018;9(1):5101 15 Guyot N et al.: Unopposed cathepsin G, neutrophil elastase, and proteinase 3 cause severe lung damage and emphysema. Am J Pathol 2014; 184(8):2197-2210 16 Alam S et al.: Oxidation of Z α1-antitrypsin by cigarette smoke induces polymerization. Am J Respir Cell Mol Biol 2011; 45(2):261-9 17 Stoller JK et al.: Delay in diagnosis of α1-antitrypsin deficiency. Chest 2005;128(4):1989-94 18 Huber F et al.: Alpha1-Antitrypsin-Mangel in Österreich: Auswertung der österreichischen Datenbank des internationalen Alpha1-Antitrypsin Registers. Wien Klin Wochenschr 2010; 122(13-14):390-6 19 Wall M et al.: Long-term follow-up of a cohort of children with alpha-1-antitrypsin deficiency. J Pediatr 1990; 116(2):248-51 20 Tejwani V et al.: The impact of delayed diagnosis of alpha-1 antitrypsin deficiency: the association between diagnostic delay and worsened clinical status. Respir Care 2019; 64(8):915-22 21 American Thoracic Society, European Respiratory Society: American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168(7):818-900 22 Sandhaus RA et al.: The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis 2016;3(3):668-82 23 Miravitlles M et al.: European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α1-antitrypsin deficiency. Eur Respir J 2017; 50(5):1700610 24 Soriano JB et al.: Trends of testing for and diagnosis of α1-antitrypsin deficiency in the UK: more testing is needed. Eur Respir J 2018; 52(1): 1800360 25 Greulich T et al.: European screening for alpha-1 antitrypsin deficiency in subjects with lung disease. Clin Respir J 2017; 11(1):90-7 26 Horváth I et al.: Diagnosis and management of α1-antitrypsin deficiency in Europe: an expert survey. ERJ Open Res 2019; 5(1):00171-2018 27 Brantly M et al.: Detection of alpha-1 antitrypsin deficiency: the past, present and future. Orphanet J Rare Dis 2020;15(1):96 28 Quinn M et al.: Obstacles to early diagnosis and treatment of alpha-1 antitrypsin deficiency: current perspectives. Ther Clin Risk Manag 2020; 16:1243-55 29 Nakanishi T et al.: The undiagnosed disease burden associated with alpha-1 antitrypsin deficiency genotypes. Eur Respir J 2020; 56(6): 2001441 30 Tanash HA et al.: Survival in individuals with severe alpha 1-antitrypsin deficiency (PiZZ) in comparison to a general population with known smoking habits. Eur Respir J 2017; 50(3): 1700198 31 Chapman KR et al.: Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet 2015; 386(9991): 360-8 32 Gurevich S et al.: Improving screening for alpha-1 antitrypsin deficiency with direct testing in the pulmonary function testing laboratory. Chronic Obstr Pulm Dis J 2021; 8(2):190-7 33 Gupta N et al.: Granularity of SERPINA1 alleles by DNA sequencing in CanCOLD. Eur Respir J 2020; 56(4):2000958
Das könnte Sie auch interessieren:

Mukoviszidose – eine Erkrankung mit Prädisposition für Pilzinfektionen
Pilzinfektionen stellen eine zunehmende Herausforderung in der Behandlung von Menschen mit Mukoviszidose (zystische Fibrose) dar. Spezifische diagnostische Schritte und therapeutische ...
Sarkoidose – Update 2025
Die Sarkoidose ist eine komplexe Multiorganerkrankung mit teils unzureichender wissenschaftlicher Evidenz. Der interdisziplinäre Austausch ist angesichts der vielen möglichen ...