Hypophosphatämie und Deformität: multidisziplinäres Management
Hypophosphatämie stellt ein Leitsymptom unterschiedlicher seltener Erkrankungen dar und wird häufig in Zusammenhang mit knöchernen Deformitäten der unteren Extremität diagnostiziert. Zur optimalen medizinischen Versorgung ist ein multidisziplinäres Setting notwendig. Die an die zugrunde liegende Erkrankung angepasste pharmakologische Therapie ist entscheidend für den Erfolg von orthopädischen Maßnahmen. Sie soll Wachstum optimieren, Beindeformitäten vorbeugen, optimale Knochenqualität intraoperativ sichern und vor Rezidiven schützen.
Keypoints
-
Beindeformität mit Hypophosphatämie stellt eine hochauffällige Kombination von Symptomen dar, die einer weiterführenden Diagnostik bedarf.
-
Viele der seltenen Erkrankungen mit dem Leitsymptom Hypophosphatämie sind medikamentös gut behandelbar.
-
Chirurgischen Verfahren zur Beinachsenkorrektur müssen zwingend multidisziplinäre Abklärung und Therapiekonzepte vorangehen.
-
Eine spezialisierte Betreuung in einem multidisziplinären Setting steht im Mittelpunkt der Behandlung.
Beindeformitäten bei Kleinkindern sind ein typischer Grund für die Erstvorstellung in der orthopädischen Praxis. Häufig liegen eine varische Achsfehlstellung (O-Bein) und Einwärtsgang mit Stolperneigung vor. Die Abgrenzung vom Normbereich zur Pathologie kann bei ausgeprägten Fällen auch für erfahrene Kinderorthopäd:innen schwierig sein, sodass im Zweifel eine weitere Abklärung des Knochenstoffwechsels sinnvoll erscheint. Bei laborchemischen Auffälligkeiten (z.B. Hypophosphatämie) und vor allem bei einer Kombination der progredienten Beindeformität mit Schmerzen ist eine multidisziplinäre Anbindung angeraten.
Unterschiedliche Erkrankungen können mit den Leitsymptomen von Beindeformitäten und Hypophosphatämie einhergehen. Mag der Begriff der Hypophosphatämie im ersten Moment für die behandelnde Orthopädie nicht hochrelevant klingen, steckt, vor allem in der Kombination mit muskuloskelettalen Beschwerden, häufig ein komplexer Pathomechanismus dahinter. Die Interpretation von Phosphatwerten im Kindesalter darf nur anhand der Referenzwerte der jeweiligen Altersgruppe erfolgen, da die Normwerte je nach Alter stark variieren. Aufgrund der fehlenden Verpflichtung, alterspezifische Referenzwerte anzugeben, sind im Zweifelsfall ein Vergleich mit verfügbaren Normwerten bzw. die Rücksprache mit dem Labor entscheidend für eine frühe Diagnose.
Knochenstoffwechselerkrankungen, die zur Hypophosphatämie führen oder damit einhergehen, sind unter anderem nutritive Rachitis, Vitamin-D-unabhängige Rachitiden, wie der Phosphatdiabetes (XLH), und seltene, mit der XLH verwandte Phosphatverlusterkrankungen. Extrem selten kann auch eine tumorinduzierte Osteomalazie (TIO) zu einer Hypophosphatämie führen. Hier entsteht durch einen FGF23 produzierenden Tumor eine hypophosphatämische Rachitis.
Es lassen sich also ernährungsbedingte von genetisch bedingten und erworbenen Rachitiden als Ursache für die Hypophosphatämie abgrenzen.
Die regionale Häufigkeit ist besonders bei der nutritiven (meist Vitamin-D-Mangel-bedingten) Rachitis sehr unterschiedlich. In Österreich ist diese aufgrund der Vitamin-D-Prophylaxe selten, aber die Dunkelziffer liegt vermutlich höher als angenommen. Die muskuloskelettalen Beschwerden und der Leidensdruck können auch bei „banaler“ Vitamin-D-Mangel-Rachitis starke Ausmaße annehmen und bereits im frühen Kindesalter zur Entstehung schwerer Beindeformitäten und Schmerzen führen. Im Vordergrund stehen der Vitamin-D-Mangel sowie erhöhte Parathormon- und Alkalische-Phosphatasespiegel als Zeichen der Gegenregulation. Hypophosphatämie und Hypokalziämie treten bei nutritiver Rachitis in weiterer Folge als Zeichen der Dekompensation auf.
Die X-chromosomale Hypophosphatämie (Phosphatdiabetes, XLH, OMIM 307800) ist eine seltene Knochenerkrankung, die durch Mutationen in PHEX („phosphate regulating endopeptidase X-linked“) verursacht wird. Während die Hypophosphatämie als Leitsymptom namensgebend für diese genetische Erkrankung ist, zeichnet sich die Erkrankung durch eine Vielzahl an weiteren klinischen Symptomen aus, die großteils das muskuloskelettale System betreffen. Komplexe Deformitäten der unteren Extremitäten und knöcherne Veränderungen zählen zu den orthopädischen Hauptmanifestationen.
„Bone clearance“ und Medikation
Sofern kein medizinischer Notfall vorliegt, ist vor orthopädischer chirurgischer Intervention eine medikamentöse Optimierung des Knochenstoffwechsels unabdingbar. Erst nach optimaler Einstellung und Freigabe durch das behandelnde endokrinologische Team („bone clearance“) ist eine orthopädische Intervention in bestmöglicher Knochenstoffwechsellage sinnvoll.
Die operativ-orthopädische Versorgung der Vitamin-D-Mangel-Rachitis gilt es besonders gut vorzubereiten. Präoperativ muss die Knochenstoffwechselsituation über Monate optimiert werden, um zum Beispiel Knochenheilung und Restwachstum zu ermöglichen, aber auch um das anästhesiologische Risiko (cave: schwere Entlgleisungen des Elektrolythaushalts bei nicht behandelter Rachitis möglich) zu minimieren. Da vor allem die von Gelenks- und Knochenschmerzen betroffenen Kinder gut auf die Medikation ansprechen, kann dies auch für die korrekte Operationsindikation entscheidend sein.
Für die nutritive Rachitis gilt also aus orthopädischer Sicht: Wenn keine Operationsdringlichkeit besteht, muss die Rachitis präoperativ medikamentös möglichst vollständig ausgeheilt sein. Gegen Wachstumsende kann aber ein verringertes Restwachstum zu einer relativen OP-Dringlichkeit führen (Abb. 1).
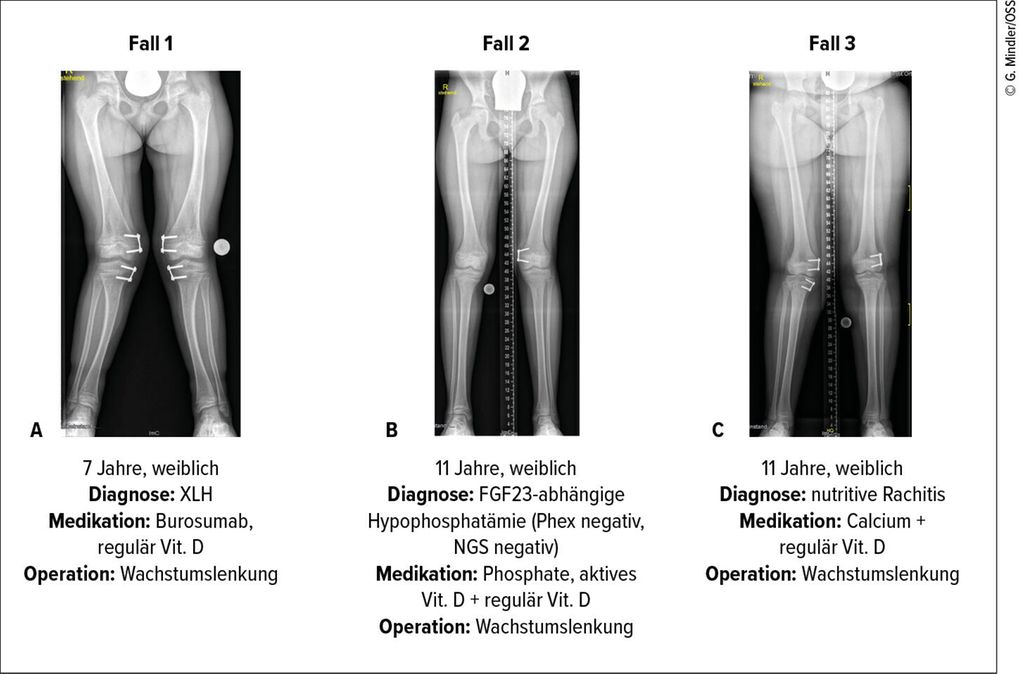
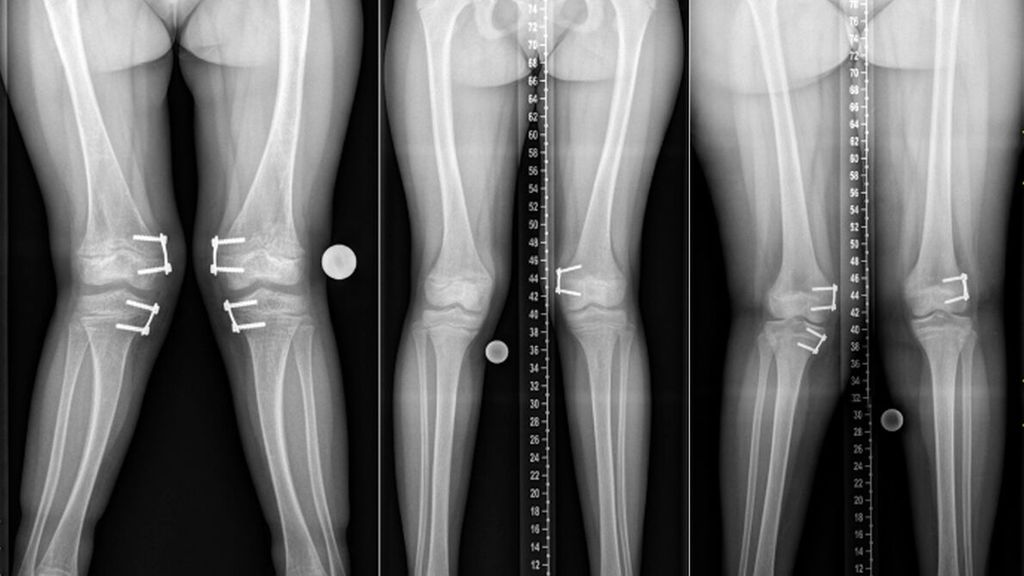
Abb. 1: Ganzbeinröntgen von Kindern mit chronischer Hypophosphatämie mit unterschiedlich stark ausgeprägten Beindeformitäten. Leitsymptome waren bei allen Patientinnen muskuloskelettale Beschwerden (Schmerz), rachitisch veränderte Wachstumsfugen (Knieröntgen), Beindeformitäten und Hypophosphatämie im Vergleich zu den Altersnormwerten. Die weitere kinderosteologische Abklärung zeigte jedoch unterschiedliche zugrunde liegende Diagnosen, welche wiederum unterschiedliche medikamentöse Therapien benötigen. Die adäquate Medikation ist entscheidend für das weitere Wachstum und letztlich für den Erfolg der chirurgischen Wachstumslenkung. In allen drei Fällen wurde bewusst noch in restrachitische Fugen operiert – zum einen, weil die medikamentöse Therapie teilweise ausgeschöpft war, zum anderen, weil aufgrund des Pubertätsfortschritts und des damit einhergehenden verminderten Wachstumspotenzials eine baldige Wachstumslenkung sinnvoll war.
Während eine gut therapierte Vitamin-D-Mangel-Rachitis eine temporäre Problematik im Wachstumsalter darstellen kann, bedürfen genetisch bedingte Hypophosphatämie-Erkrankungen einer langfristigen multidisziplinären Betreuung. Die osteologische Versorgung spielt vor allem bei XLH lebenslang eine wichtige Rolle. Bis vor Kurzem standen lediglich 1,25-OH-VitaminD und orale Phosphatsalze als medikamentöser Therapieansatz zur Verfügung. Der monoklonale Antikörper Burosumab stellt jedoch eine neue therapeutische Option dar, die zu einer signifikanten Verbesserung der rachitischen Komponente und der Hypophosphatämie bei Kindern führt.
Von neuen Therapiemöglichkeiten mit dem FGF23-neutralisierenden Antikörper Burosumab wird eine Verbesserung von Beindeformitäten im Wachstumsalter erhofft. Erste orthopädische Daten konnten diese Erwartungen bezüglich bereits aufgetretener Veränderungen der Beinachse nicht erfüllen. Dennoch erscheint es möglich, dass eine optimale präoperative Medikation mit Burosumab zu einer Verbesserung des Outcomes sowie zu einer Prävention von Progressionen oder Rezidiven führt. Das könnte entscheidend für den Erfolg von wachstumslenkenden Operationen sein. Somit gilt für die Wachstumslenkung bei XLH: Ein komplettes Ausheilen der rachitischen Veränderungen (Röntgen) muss nicht zwingend abgewartet werden, da dieses bei manchen Kindern trotz optimierter Medikation nicht eintritt.
Zusätzlich wurde eine deutlich verbesserte Heilungsrate von XLH-assoziierten Pseudofrakturen bei Erwachsenen beschrieben, sodass sich hier bereits ein Paradigmenwechsel abzeichnet, weg von einer intramedullären Stabilisierung und hin zu einer optimalen medikamentösen Therapie.
Eine gute Compliance und verbesserte Phosphatwerte schließen jedoch die Entstehung von Deformitäten an der unteren Extremität, Pseudofrakturen oder Rezidive keineswegs gänzlich aus.
Orthopädische Betreuung im Wachstumsalter (XLH)
Die kinderorthopädische Betreuung sollte bei XLH bereits im ersten Lebensjahr erfolgen und das gesamte Wachstumsalter gegeben sein. Die Hauptbeschwerden im Kindesalter sind muskuloskelettale Schmerzen, frühe Ermüdung und Beindeformitäten. Neben dem gleichzeitigen Vorliegen der Beindeformitäten in unterschiedlichen Ebenen (frontal, transversal, sagittal) ist auch der multiapikale Charakter der Deformität typisch für den XLH-Knochen.
Wachstumslenkende OP-Techniken (Hemiepiphyseodese) werden in Zentren für die Behandlung von Beindeformitäten bei Kindern mit XLH verwendet. Obwohl diese OP-Technik vergleichsweise einfach ist, verlangt die Behandlung von Kindern mit XLH mit dieser Methode eine hochgradige Spezialisierung und Erfahrung und optimierte multidisziplinäre Betreuung. Es können Deformitäten nämlich nur auf Ebene der Wachstumsfuge korrigiert werden und es bedarf eines ausreichenden Restwachstums. Bei der Korrektur in jungen Jahren muss von einer hohen Rezidivrate ausgegangen werden und es kann das Konzept der wiederholten Wachstumslenkung angewandt werden, um langfristig die Notwendigkeit invasiver Operationen zu reduzieren. Gewisse Deformitäten (Maltorsion, Procurvatum) können jedoch mit derzeitigen wachstumslenkenden OP-Verfahren nicht ausreichend korrigiert werden. Deshalb können auch bei Kindern mit XLH trotz optimaler Therapie und minimalinvasiver OP-Methoden Verfahren mit Osteotomien notwendig sein, welche zur bestmöglichen Knochenheilung gut geplant nach erfolgter „bone clearance“ durchgeführt werden.
Orthopädische Betreuung im Erwachsenenalter (XLH)
Während im Idealfall die Knochendeformitäten von XLH-Patient:innen im jungen Erwachsenenalter weitgehend korrigiert sind, kommen zusätzliche Symptome, wie schwere Enthesiopathien, chronische frakturähnliche Veränderungen (Pseudofrakturen) und frühe Arthrose, zur Ausprägung. Durch die neue Antikörpertherapie lassen sich zwar Pseudofrakturen bei Erwachsenen mit XLH teils gut und OP-frei therapieren, allerdings konnte bislang noch keine signifikante Verbesserung der Enthesiopathien und der vorzeitigen Arthrose nachgewiesen werden.
Da Enthesiopathien bei diesem Krankheitsbild meist auch orthopädisch (konservativ wie operativ) nicht sonderlich zufriedenstellend behandelt werden können, bleibt dieses Symptom auch in Zukunft eine multidisziplinäre Herausforderung (und eine Ursache des hohen Leidensdrucks der Betroffenen).
Vorzeitige Gelenksabnützungen, meist kombiniert mit ausgeprägter Deformität der unteren Extremität, bedürfen einer hohen endoprothetischen Expertise. Oftmals ist auch eine vorbereitende Achskorrektur bei besonders stark ausgeprägten Deformitäten sinnvoll, um die Implantation einer Prothese zu erleichtern und deren Lebensdauer zu verlängern. Die optimale Knochenstoffwechsellage erscheint auch für die prothetische Versorgung sinnvoll, wobei es derzeit keine aktuellen Daten hinsichtlich des endoprothetischen Outcomes im Zusammenhang mit XLH-Medikation gibt.
Für die behandelnden Orthopäd:innen ist die Kenntnis des komplexen Phänotyps bei XLH entscheidend, um eine Schmerzsymptomatik richtig einzuordnen, Operationen sicher zu planen und knöcherne Veränderungen korrekt diagnostizieren zu können (z.B. Differenzierung zwischen periprothetischer Fraktur und periprothetischer XHL-assoziierter Pseudofraktur).
Zusammenfassung
Das Vorliegen von Hypophosphatämie und Beindeformitäten soll für die behandelnde Orthopädie als „red flag“ angesehen werden. Die weitere Abklärung und Therapie der Hypophosphatämie obliegen der endokrinologischen Fachexpertise. Orthopädische Operationen sollten im multidisziplinären Setting und nach endokrinologischer Freigabe („bone clearance“) geplant werden.
Literatur:
bei den Verfasser:innen
Das könnte Sie auch interessieren:

Frühe Unterwassertherapie nach totaler Hüft- und Knieendoprothese
Eine frühzeitige Durchführung der Unterwassertherapie mit wasserdichtem Folienverband ab dem vierten postoperativen Tag ist im Allgemeinen sicher, wirksam und ohne Nebenwirkungen.
Aktuelles zur anteromedialen Knieinstabilität
Kombinierte Verletzungen des medialen Kollateral- (MCL) und des vorderen Kreuzbandes (VKB) zählen zu den häufigsten multiligamentären Knieverletzungen. Bei vermeintlich isolierten VKB- ...
Arthroskopische Behandlung von Glenoidrandfrakturen
In der Literatur werden die Begriffe knöcherne Bankart-Läsion und Glenoidrandfraktur häufig synonym verwendet, obwohl sie sich hinsichtlich Pathomechanismus und Therapie deutlich ...