
Neue Gentherapien zur Behandlung der spinalen Muskelatrophie
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die spinale Muskelatrophie (SMA) ist eine schwere, lebenslimitierende neurodegenerative Erkrankung. Bisher führte sie zum vorzeitigen Versterben der Patienten in Abhängigkeit vom Schweregrad. Mittlerweile stehen vielversprechende krankheitsmodifizierende Gentherapien zur Verfügung, welche die motorischen Funktionen verbessern und die Sterblichkeit verringern.
Keypoints
-
Durch die Anwendung der Gentherapie für die SMA konnte in den letzten Jahren bereits ein erheblicher medizinischer Fortschritt zum Wohle der schwer betroffenen Patienten erzielt werden mit deutlicher Besserung des Überlebens, Stabilisierung bzw. Verbesserung der motorischen Funktion und weniger Beatmungspflichtigkeit.
-
Aktuell stehen 3 Substanzen (Onasemnogene Abeparvovec [Zolgensma®], Nusinersen [Spinraza®] und Risdiplam) mit teilweise unterschiedlichem Wirkmechanismus und Applikationsweg zur Behandlung der SMA zur Verfügung.
-
Obwohl generell die Wirksamkeit der Gentherapien umso besser ist, je früher die Patienten behandelt werden, legen die Ergebnisse der Studien und Verlaufsbeobachtungen offen, dass auch jugendliche und erwachsene SMA-Patienten von einer Therapie profitieren können und dass sich nicht nur eine Verschlechterung verhindern lässt, sondern auch bei zumindest einem Teil der Erkrankten eine klinisch bedeutsame Besserung erzielt werden kann.
Die spinale Muskelatrophie (SMA) ist eine autosomal-rezessiv neuromuskuläre Erbkrankheit. Bei etwa 95% aller betroffenen Patienten liegt eine homozygote Deletion u.a. des Exons 7 des sogenannten Survival-Motor-Neuron-1-Gens (SMN-1) vor. Der Verlust des SMN1-Genproduktes ist der zentrale Pathomechanismus der Erkrankung und zieht einen fortschreitenden Untergang der Vorderhornzellen (α-Motoneuronen) im Rückenmark und Hirnstamm nach sich. Die daraus resultierende Muskelatrophie führt zu den Leitsymptomen der Erkrankung, einer progredienten proximalen Muskelschwäche und Muskelatrophie mit Verlust der Muskeleigenreflexe. Eine zunehmende respiratorische Insuffizienz mit Notwendigkeit der non-invasiven Beatmung ist letzten Endes der lebenslimitierende Faktor. Das SMN2-Gen ist bis auf eine C>T-Substitution auf Exon 7 mit dem SMN1-Gen identisch. Diese Substitution führt jedoch dazu, dass ein mehrheitlich verkürztes, nicht funktionsfähiges SMN-Protein produziert wird. Die Anzahl der SMN2-Genkopien ist variabel und beeinflusst maßgeblich den Schweregrad der Erkrankung. Die SMA wird entsprechend dieser Schweregrade in drei unterschiedliche Formen eingeteilt (Typ I–III) (Tab.1).

Tab. 1: Bei der SMA werden je nach Schweregrad drei verschiedene Typen unterschieden
Aufgrund dieser genetischen Besonderheiten gibt es verschiedene gentherapeutische Ansätze (Abb.1). Bei der Genersatztherapie wird über eine einmalige intravenöse Verabreichung das funktionale SMN1-Gen mittels Virusvektoren in die Zielzellen eingebracht. Ein weiterer Ansatz zur Behandlung der SMA ist die Applikation von Antisense-Oligonukleotiden (ASO) oder sogenannter „small molecules“, die das Spleißen des SMN2-Gens beeinflussen und damit zu einer vermehrten Produktion des SMN-Proteins führen. Alle bisher verfügbaren Daten zur medikamentösen Therapie der SMA zeigen übereinstimmend, dass der therapeutische Nutzen vor allem vom Krankheitsstadium und vom Lebensalter bei Therapiebeginn abhängt.
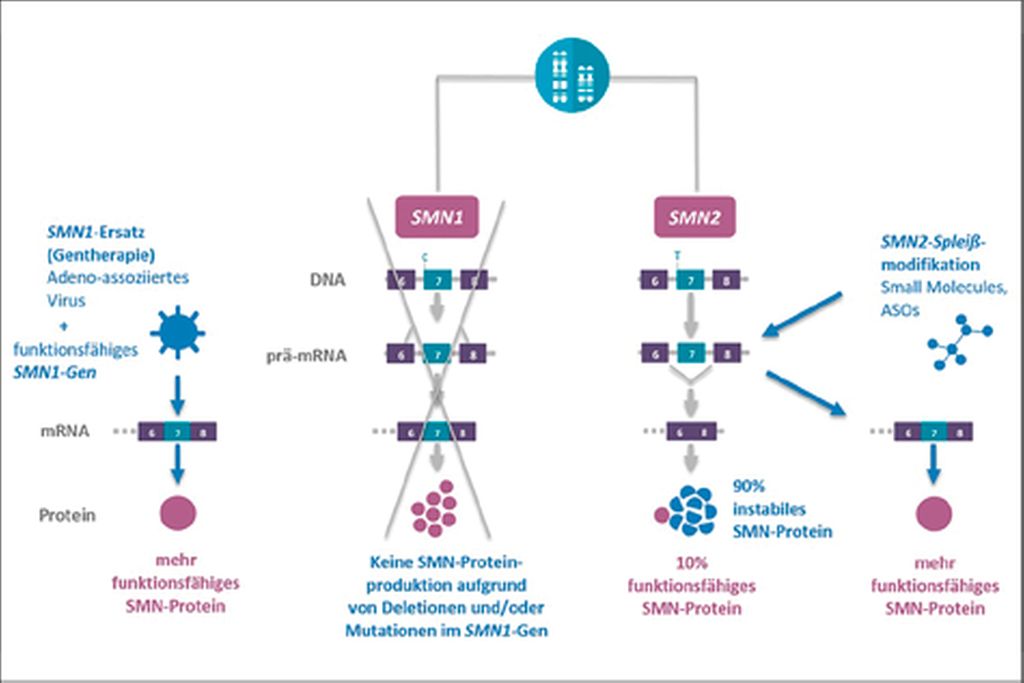
Abb. 1: SMN1-Ersatz und SMN2-Spleißmodifikation
Genersatztherapie
Onasemnogene Abeparvovec (Zolgensma®) ist ein Genersatztherapeutikum für die kausale Therapie der 5q-assoziierten SMA (mit einer biallelischen Mutation des
SMN1-Gens und einer klinischen Diagnose einer SMA Typ 1 sowie einer biallelischen Mutation des SMN1-Gens und bis zu drei Kopien des SMN2-Gens). Mittels eines intravenös applizierten Adenovirus-assoziierten Vektors (AAV) wird eine funktionsfähige Kopie des SMN1-Gens in den Zellkern der α-Motoneuronen eingeschleust und die Funktion des defekten SMN1-Gens ersetzt. Auf diese Weise kann sehr schnell nach der Gabe bereits funktionsfähiges SMN-Protein gebildet werden (Abb.1). Der Serotyp des Virusvektors (AAV9) zeichnet sich durch die Fähigkeit aus, die Blut-Hirn-Schranke zu überwinden und damit ins Gehirn und Rückenmark zu gelangen. Der AAV9-Vektor wird dabei so modifiziert, dass dieser sich nicht in das Wirtsgenom des Menschen integriert. Neben der Wirkung in der Zielzelle müssen im kurzfristigen Intervall nach Gabe vor allem sogenannte „Off-target“-Effekte berücksichtigt und regelmäßig überwacht werden. Sie entstehen in erster Linie durch Ansteuerung weiterer Gewebe wie der Leber (Transaminasenerhöhung), des Herzens (Myokarditis mit Erhöhung von Troponinwerten) und des blutbildenden Systems (Blutbildveränderungen, insbesondere Thrombopenie). Aufgrund der Antikörperbildung gegen AAV kann Zolgensma® nur einmalig appliziert werden. Die Verabreichung der Therapie erfolgt nach Ausschluss potenzieller Kontraindikationen stationär unter einer fortlaufenden Steroidtherapie, welche 24 Stunden vor der Gabe begonnen und für mindestens 4 Wochen durchgeführt und langsam ausgeschlichen wird.
In Anlehnung an das Konsensuspapier der deutschen Vertretung der Gesellschaft für Neuropädiatrie (GNP) und der deutschen Behandlungszentren unter Mitwirkung des Medizinisch-Wissenschaftlichen Beirates der Deutschen Gesellschaft für Muskelkranke (DGM) werden zur besseren Übersicht drei Phasen der spezialisierten Betreuung unterschieden. Die prästationäre Phase beinhaltet eine entsprechende Vorbereitung, Indikationsstellung und Beantragung der Gentherapie. Im Rahmen der stationären Phase erfolgen die tatsächliche Applikation und die entsprechende engmaschige Observation. Die wichtigste Phase ist die poststationäre Phase, welche die Langzeitbeobachtung nach erfolgter Gentherapie umfasst. Die Besonderheiten der Therapie mit Zolgensma® umfassen unter anderem ein heterogenes Patientenkollektiv (Neugeborene, Säuglinge und Kleinkinder mit einem großen Spektrum an klinischer Symptomatik und Erkrankungsverlauf), nur begrenzte weltweite Erfahrungswerte sowie ein langfristiges Monitoring für unerwartete Nebenwirkungen im Verlauf und stellen besonders hohe Ansprüche und Anforderungen an die Struktur und die personelle Ausstattung der behandelnden Kliniken dar. Zolgensma® wurde aufgrund der guten Wirksamkeit in einer offenen Studie (START) an symptomatischen SMA-1-Kindern, die vor dem 6. Lebensmonat behandelt worden waren, von der EMA am 18.5.2020 zugelassen. Die Extensionsstudie sowie Kongressberichte zeigen, dass die gute Wirksamkeit in vielen Fällen über einen längeren Zeitraum erhalten bleibt. Vorläufige Daten einer Studie an präsymptomatischen SMA-Kindern mit 2 oder 3 SMN2-Allelen (SPRINT) sind sehr vielversprechend und können vereinzelt eine annähernd normale motorische Entwicklung aufzeigen. Alle bisher verfügbaren Daten belegen, dass der therapeutische Nutzen vor allem vom Lebensalter bei Therapieinitiierung und vom Krankheitsstadium abhängt. Ein Therapiebeginn in der präsymptomatischen Phase ist besonders vielversprechend. Insgesamt hat die Genersatztherapie der SMA mit Zolgensma® das Potenzial, den Krankheitsverlauf signifikant und positiv zu beeinflussen.
Spleißmodifikation am SMN2-Gen durch molekulare Therapien
Das Antisense-Oligonukleotid (ASO) Nusinersen (Spinraza®) wurde in Europa im Juli 2017 als erste krankheitsmodifizierende Therapie der SMA für alle Subtypen und Altersstufen zugelassen. Es handelt sich hierbei um modifizierte einzelsträngige RNA-Moleküle, welche an das komplementäre Intron 7 der SMN2-prä-mRNA binden und somit das Entfernen des Exons 7 durch das Spliceosom verhindern. Das Resultat ist eine Hochregulation des funktionsfähigen SMN2-Proteins (Abb.1). Die Behandlung sollte so früh wie möglich nach Diagnosestellung mit 4 Aufsättigungsdosen begonnen werden. Die Applikation erfolgt per Lumbalpunktion direkt in den Liquorraum. Anschließend wird alle 4 Monate eine Erhaltungstherapie verabreicht. Die Zulassung basiert auf den beiden randomisierten Doppelblindstudien ENDEAR, in welcher symptomatische SMA-1-Kinder vor dem 7. Lebensmonat behandelt wurden, und CHERISH, in der SMA-2- und SMA-3-Kinder zwischen dem 2. und 9. Lebensjahr eingeschlossen wurden. In der NURTURE-Studie wurden präsymptomatische SMA-Kinder mit 2 oder 3 SMN2-Allelen behandelt. Auch hier zeigten sich dramatische Effekte auf Überleben, Beatmungsnotwendigkeit und motorische Entwicklung.
Nusinersen hat ein günstiges Sicherheitsprofil. Die häufigsten Nebenwirkungen wie Infektionen der oberen und unteren Atemwege sowie Obstipation standen entweder im Zusammenhang mit der Grunderkrankung oder waren der Lumbalpunktion zuzuschreiben. Seit Kurzem liegen auch Daten aus mehreren offenen Beobachtungsstudien zur Wirksamkeit von Nusinersen in der Behandlung erwachsener SMA-Patienten vor. Nach 14 Monaten Beobachtungszeit konnten bereits klinisch bedeutsame positive Effekte von Nusinersen in 40–69% der adulten SMA-Patienten nachgewiesen werden. Diese Effekte waren abhängig von der Schwere der Erkrankung. Bei gehfähigen, milder Betroffenen sowie bei SMA-3-Patienten war die Wirkung am größten. Das Alter hingegen spielte keine Rolle.
Bei Risdiplam, einem „small molecule“, handelt es sich um ein einzelsträngiges Oligonukleotid, das ähnlich wie Nusinersen das Spleißen von SMN2 beeinflusst und zur Bildung von mehr funktionsfähigem SMN-Protein führt. Es ist jedoch in der Lage, die Blut-Hirn-Schranke zu überwinden, wodurch eine orale Aufnahme ermöglicht wird. Im Jahr 2020 hat damit die US-Arzneimittelbehörde FDA erstmals ein orales Medikament unter dem Handelsnamen Evrysdi® zur Behandlung der spinalen Muskelatrophie zugelassen. Die Zulassung basiert auf einer Open-Label-Studie (FIREFISH), in der symptomatische SMA-1-Kinder vor dem 7. Lebensmonat behandelt wurden, und einer doppelblinden, placebokontrollierten Studie (SUNFISH), in welcher SMA-2- und SMA-3-Patienten zwischen dem 2. und 25. Lebensjahr eingeschlossen wurden. Wie auch bei anderen Substanzen können eine Stabilisierung bzw. eine Verbesserung der motorischen Funktionen und eine Verlängerung der beatmungsfreien Überlebenszeit bei frühem Therapiebeginn erzielt werden. Erfreulicherweise zeigen die Subgruppenanalysen der SUNFISH-Studie von 18–25-Jährigen ebenso einen Therapieeffekt im Sinne einer Verbesserung der Selbstständigkeit. Weitere Studien mit anderen Altersgruppen und Patientenpopulationen werden noch durchgeführt: JEWELFISH – eine offene explorative Studie mit Patienten im Alter von 6 Monaten bis 60 Jahren mit SMA Typ II oder III, die zuvor mit einer anderen SMN-Targeting-Therapie behandelt wurden, und RAINBOWFISH – eine Studie für Patienten mit präsymptomatischer SMA. Eine Zulassung durch die EMA wird demnächst erwartet.
Literatur:
bei den Verfassern
Das könnte Sie auch interessieren:
Angepasste Therapien und Biomarker verbessern den Krankheitsverlauf bei MS
Neue Biomarker und sensitivere Analysemethoden erleichtern die Behandlungsauswahl bei Multipler Sklerose und bilden den Krankheitsverlauf unter den Therapien immer verlässlicher ab. Auf ...

Interdisziplinäre Therapie der intrazerebralen Blutung
Aktuelle Studienergebnisse brachten erstmals einen positiven Effekt operativer Therapieverfahren auf das funktionelle Outcome bei Patient:innen mit intrazerebraler Blutung. Für die ...

Wenn das Sprechen schwerfällt – Dysarthrien verstehen und behandeln
Dysarthrien sind erworbene neurogene Störungen der Sprechmotorik, die die Ausführung und Koordination der für das Sprechen benötigten Bewegungen beeinträchtigen. Neben bekannten, ...