
Behandlung von Gliomen im Erwachsenenalter
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Mit der Veröffentlichung der 5. Überarbeitung der WHO-Klassifikation der Tumoren des zentralen Nervensystems vor gut einem Jahr wurde die Notwendigkeit, die Behandlungskonzepte zu aktualisieren, in den Vordergrund gerückt.1 Die Fachgesellschaften waren schon sehr früh aktiv. So wurden die Leitlinien zur Behandlung von Gliomen im Erwachsenenalter der Europäischen Neuroonkologischen Gesellschaft (EANO) bereits Ende 2020,2 die der deutschen Gesellschaft für Neurologie im Jahr 2021 aktualisiert.3
Keypoints
-
Die neuen molekulardiagnostischen Algorithmen zur Klassifikation von Gliomen in Anlehnung an die 5. WHO-Klassifikation der Hirntumoren sind ein Standard oder sollen es werden.
-
Prognose und Therapieansprechen sind damit präziser vorhersagbar.
-
Subgruppen profitieren von der Präzisionsmedizin (zielgerichtete Therapien).
-
Experimentelle Behandlungsverfahren sollten bei Gliomen bevorzugt in klinischen Studien eingesetzt werden (Primärtherapie und in der Rezidivsituation).
Auch die bereits abgeschlossenen und laufenden großen klinischen Phase-II/III-Studien musste man im Licht der neuen Erkenntnisse interpretieren und unter Berücksichtigung aktueller Klassifikationen neue klinische Studien mit innovativen Ansätzen konzipieren (z.B. eine internationale, nahtlose „Response Adaptive Randomization Platform“-Phase-II/III-Studie zur Bewertung mehrerer Therapien bei neu diagnostiziertem und rezidivierendem GB, kurz „GB AGILE: Global Adaptive Trial Master Protocol“).
Was gibt es Neues?
Die 5. Edition der WHO-Klassifikation 2021, mit nun >120 Tumortypen, beinhaltet neue Tumortypen, Subtypen und Gradierung (in arabischen Ziffern) und stellt die Kliniker vor eine besondere Herausforderung. Neben histologischen und molekularen Kriterien werden auch die Empfehlungen des Konsortiums c-IMPACT-NOW* berücksichtigt. Basierend auf den Empfehlungen dieses Konsortiums wurde bei IDH-Wildtyp-Gliomen ohne klassische Merkmale eines Grad-4-Tumors, wie Nekrosen und/oder das Vorhandensein mikrovaskulärer Proliferate, ein Glioblastom molekular definiert durch Vorhandensein einer EGFR-Amplifikation, einer TERT-Promoter-Mutation oder Zugewinnen auf Chromosom 7 bei Verlust von Chromosom 10 (+7/–10).
Empfohlen wird, dass neben IDH(Isocitrat-Dehydrogenase)-Mutations-Status 1p/19q-Kodeletion und Histon-H3-Mutation in der Diagnostik herangezogen werden4 und in die Therapieentscheidungen einfließen. Insbesondere die epigenetischen Merkmale wie MGMT-Promotermethylierung können bei Glioblastomen, IDH-Wildtyp, für den Einschluss in klinische Studien und bei älteren Betroffenen (arbiträre Grenze 65–70 Jahre) maßgeblich zur klinischen Therapieentscheidung herangezogen werden. Die neue Klassifikation führte sowohl zur Reklassifikation einiger Tumorentitäten (Oligoastrozytome werden je nach molekularen Markern entweder Astrozytomen oder Oligodendrogliomen zugeordnet; Gliomatosis cerebri ist keine eigenständige Tumorentität mehr, lediglich ein Wachstumsmuster; IDH-mutiertes Astrozytom, ZNS-WHO-Grad 4, ersetzt den Terminus sekundäres IDH-mutiertes Glioblastom) als auch zu neuen, rein molekular definierten Tumorentitäten wie zum Beispiel hochgradiges Astrozytom mit piloiden Merkmalen, die nur durch das Mikroarray-basierte DNA-Methylierungsprofil diagnostiziert werden können. Die DNA- Methylierungsanalyse erlaubt auch eine verbesserte diagnostische Subgruppierung in histologisch oder molekularpathologisch unklaren Fällen.5,6
Zudem lassen sich neue Tumorentitäten abgrenzen, für die das beste therapeutische Vorgehen und prognostische Merkmale noch definiert werden müssen. Für die neuen Entitäten, insbesondere die H3-Alteration, ist die Behandlung im Rahmen klinischer Studien oder prospektiver Register wünschenswert, um so die neuen Therapiestandards rasch generieren zu können.7,8
Veränderungen der einzelnen Histone definieren zwei neue Tumortypen der IDH-Wildtyp-Gliome: das Histon-3-K27M-veränderte diffuse Mittelliniengliom (Lokalisation: Thalamus, Pons, Hirnstamm und Rückenmark) und das Histon-3.3-G34-mutierte diffuse hemisphärische Gliom. Beide kommen im Adoleszenten- und jungen Erwachsenenalter vor.
Verständnis der genetischen und epigenetischen Alterationen (Beispiele: IDH-1- und -2-Mutation, MGMT**-Promotor-Methylierung, Verlust der Histon-H3-Trimethylierung) hat das Potenzial, die Prognose besser vorherzusagen, das Ansprechen auf die Therapie genauer abzuschätzen und grundsätzliche tumorbiologische Mechanismen so zu verstehen, dass sich daraus neue Therapien weiterentwickeln lassen.
Therapie diffuser Gliome
Die dichotome Unterteilung in IDH-Wildtyp-Gliome (=Glioblastom) ZNS-WHO-Grad 4 und IDH-mutierte Gliome des Erwachsenenalters ZNS-WHO-Grad 2–4 deutet auf unterschiedliche Tumorbiologie und klinische Verläufe dieser Tumoren hin (Tab. 1). Diese werden insbesondere von den pädiatrischen Tumortypen scharf abgegrenzt, da sich die Entitäten stark in den Verläufen und der Prognose unterscheiden. Die IDH-mutierten diffusen Gliome haben unabhängig von der Gradierung einen längeren natürlichen Krankheitsverlauf und eine günstigere Prognose als die IDH-Wildtyp-Tumoren. Ein molekularer Nachweis einer Homozygoten-Deletion des Genlocus für die zyklinabhängigen Kinasen A und 2B (CDKN2A/B) stellt erstmals einen Biomarker dar, der mit einer deutlich schlechteren Prognose bei Patienten mit IDH-mutierten Astrozytomen und Oligodendrogliomen assoziiert ist und in den diagnostischen Algorithmus als Indikator für Astrozytome ZNS-WHO-Grad 4 eingeflochten wurde.9,10 Die Wahl der tumorspezifischen Therapie soll immer interdisziplinär getroffen werden.
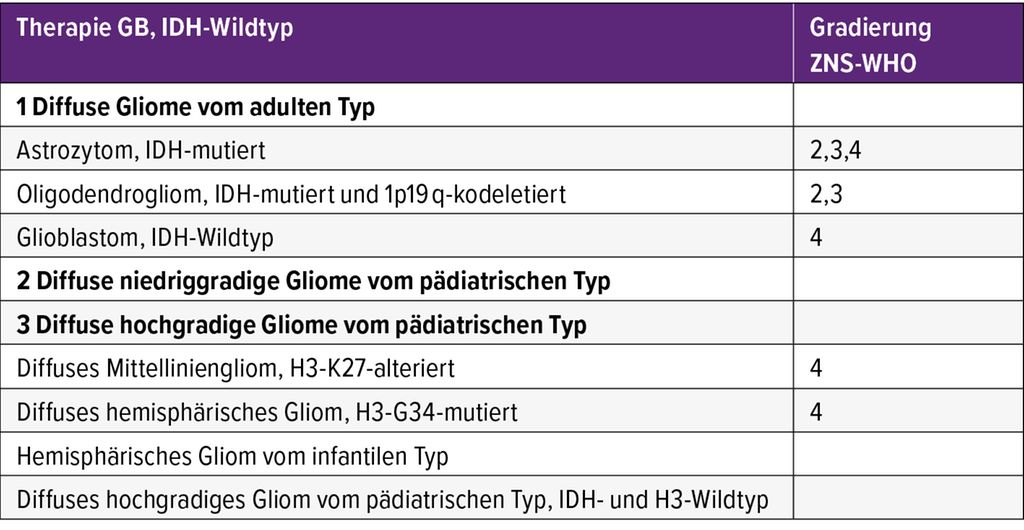
Tab. 1: Einteilung diffuser Gliome gemäß der neuen WHO-Klassifikation der Tumoren des ZNS 2021
Glioblastom, IDH-Wildtyp
Bei neu diagnostiziertem Glioblastom bleibt die Standardtherapie gestützt auf 4 Säulen:
-
Maximal mögliche Tumorresektion unter Schonung funktioneller Areale ohne Produktion neuer neurologischer Defizite, ansonsten Biopsie zur Diagnosesicherung,
-
Strahlentherapie (Zieldosis 60 Gy) in Kombination mit Temozolomid(TMZ)-Chemotherapie, das sogenannte „Stupp-Schema“,11 gefolgt von
-
6 Zyklen Erhaltungs-TMZ-Monotherapie. Auch eine Kombinationstherapie mit Lomustin und Temozolomid bei methyliertem GB und guter Knochenmarksreserve ist in der Primärtherapie möglich.12
-
Einsatz elektrischer Wechselfelder – TTF („Tumor Treating Fields“)13 – nach abgeschlossener Radiochemotherapie ist die vierte Therapiemodalität, die in der Primärtherapie des Glioblastoms angeboten werden sollte, da sie das PFS und OS verlängert.
Bei älteren Patienten können in Abhängigkeit vom Karnovsky-Index und von Komorbiditäten eine hypofraktionierte Strahlentherapie (15x) bei unmethyliertem Glioblastom oder nur Temozolomid-Chemotherapie bei methyliertem GB angeboten werden.14 Der Einschluss in klinische Studien wird von allen internationalen Leitlinien zur Behandlung des IDH-Wildtyp-Glioblastoms mit unmethyliertem MGMT-Promotor entlang der gesamten Behandlungstrajektorie empfohlen.
IDH-mutierte diffuse Gliome
Bei den „niedrigergradigen Gliomen“, wo die Grenzen der Gradierung zwischen ZNS-WHO-Grad 2 und 3 immer fließender werden, haben mehrere Faktoren Einfluss auf den Zeitpunkt und die Reihenfolge der tumorspezifischen Therapie. Die Wahl der Therapie soll nach dem Konzept des „shared decision-making“ mit dem Patienten getroffen werden. Bei Vorhandensein von positiven prädiktiven Faktoren wie möglichst vollständiger Tumorresektion und jungem Alter (<40 Jahren) kann die Therapie bis zur radiografischen oder symptomatischen Manifestation postponiert werden.3
Umgekehrt sollen Betroffene mit hohem Risiko, definiert durch subtotale Resektion (>1cm Residualtumor im MRT), Alter >40 Jahre, Tumorgröße >4–6cm, mittellinien-überschreitende Tumoren, therapierefraktäre epileptische Anfälle und prächirurgische neurologische Defizite, frühzeitig einer Therapie zugeführt werden (Abb. 1).
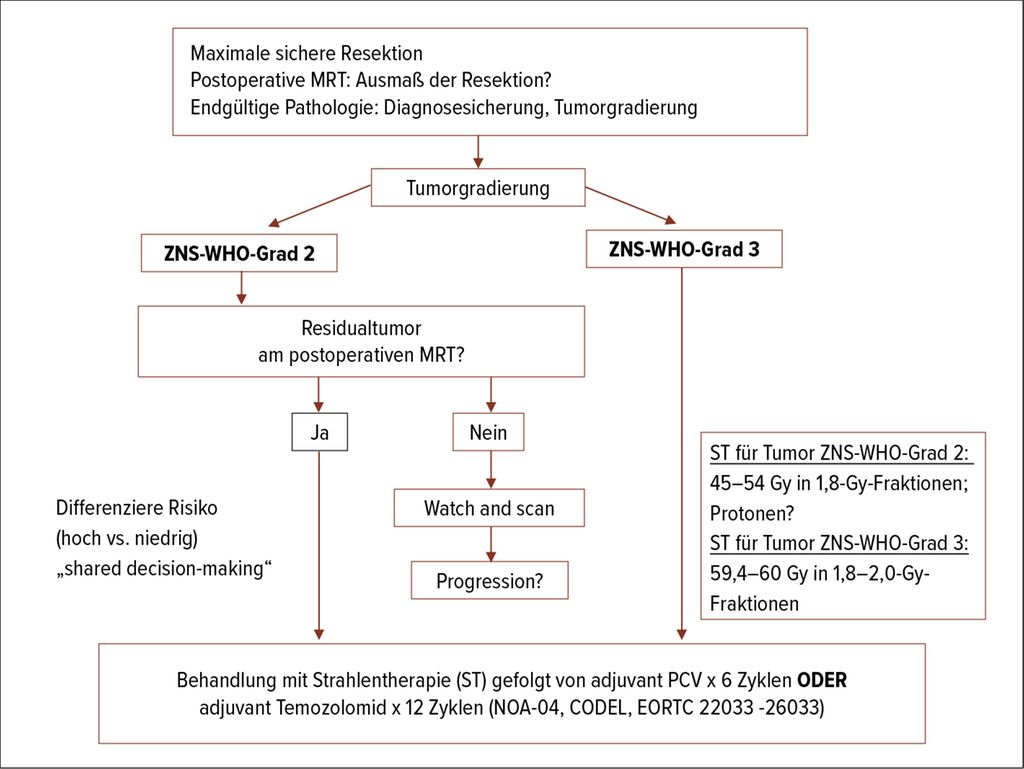
Abb. 1: Pfad der Therapiewahl bei Tumoren ZNS-WHO-Grad 2 und -Grad 3
Die Behandlung der Oligodendrogliome, IDH-mutiert, 1p19q-kodeletiert, ZNS-WHO-Grad 2, sowie der Astrozytome, IDH-mutiert, ZNS-WHO Grad 2, besteht nach der mikrochirurgischen Resektion aus Hochpräzisionsstrahlentherapie gefolgt von adjuvanter Chemotherapie mit 4–6 Zyklen Procarbazin, Lomustin, Vincristin (PCV) oder 12 Zyklen Temozolomid. Auch wenn bisher keine randomisierten Studien zum Direktvergleich von PCV und Temozolomid bestehen, wird aufgrund des günstigen Nebenwirkungsprofils die Temozolomid-Therapie bevorzugt eingesetzt.
Bei Grad-3-Gliomen soll unabhängig vom Ausmaß der Resektion eine adjuvante Therapie angeboten werden. Bereits 2017 wurde die erste Interimsanalyse der CATNON-Studie veröffentlicht. In dieser konnte gezeigt werden, dass nach einer Strahlentherapie 12 Zyklen von Temozolomid-Erhaltungstherapie bei IDH-mutierten Astrozytomen ZNS-WHO-Grad 3 einer Radiotherapie alleine überlegen sind (82 vs. 47 Monate; HR: 0,64, 95% CI: 0,52–0,79). Dies gilt nicht für die IDH-Wildtyp-Tumoren (diese würden nach der neuen WHO-Klassifikation der Gruppe der Glioblastome zugeordnet werden).
Die neuen Erkenntnisse der CATNON-Studie (randomisierte Phase-III-Studie bei neu diagnostizierten anaplastischen Gliomen ohne 1p19q-Kodeletion) aus dem Jahr 2022 nach 56 Monaten Nachbeobachtungszeit lassen eine untergeordnete Rolle des konkomitanten Temozolomid während der Bestrahlung bei IDH-mutierten Grad-3-Tumoren ohne 1p19q-Kodeletion vermuten.15
Extralangzeit-Überlebensdaten der beiden Studien EORTC 26951 und RTOG 9402 zu Oligodendrogliomen Grad 3 mit 18–19 Jahren Nachbeobachtungszeit wurden 2022 publiziert.16 Bei Oligodendrogliomen ZNS-WHO-Grad 3 konnte eine 40%ige Reduktion der Todesrate durch Zugabe von PCV zur RT im Vergleich zu RT erzielt werden. Das mediane Gesamtüberleben wurde mit ca. 14 Jahren angegeben und die geschätzten PFS- und OS-Wahrscheinlichkeiten nach 20 Jahren ab Randomisierung wurden mit 30 und 35% berechnet.
Das unterstreicht noch einmal die Notwendigkeit einer adäquaten Therapiewahl zur Verhinderung relevanter früher und später kognitiver Defizite bei dieser Tumorentität. Eine „neurokognitive Basisbeurteilung“ kann im Verlauf zur Objektivierung der Beschwerden eingesetzt werden, um den Betroffenen die Stärken und Schwächen aufzuzeigen und daraus realistische Ziele für eine kognitive Rehabilitation abzuleiten.
IDH-mutierte Grad-4-Gliome treten im Vergleich zu IDH-Wildtyp-Tumoren häufiger bei jüngeren Betroffenen auf und sind öfters MGMT-methyliert. Sie werden entweder analog zum Glioblastom oder in Anlehnung an die CATNON-Studie behandelt.
Neue Substanzklasse – IDH-Inhibitoren
Mutationen der Isocitrat-Dehydrogenase 1 führen zur Produktion des Onkometaboliten 2-Hydroxyglutarat (2-HG). IDH-1- (weniger IDH-2-) Mutationen sind häufig bei niedrigergradigen Gliomen und führen zu epigenetischen und genetischen Veränderungen, die die Onkogenese fördern.
Epigenetik
Epigenetik reguliert die Genaktivität und beginnt mit DNA und Histonen, die sich funktionell im Chromatin vereinen. Histone sind Protein-Oktamere, um die sich die DNA wickelt und so dicht verpackt wird. DNA- und Histon-Modifizierungen erfolgen durch Anhängen bestimmter Gruppen (Methylgruppen oder Acetylgruppen) an die DNA-Basen oder Histone und dadurch können das Ablesen der Information und so die Funktion gesteuert werden. So entsteht eine zweite Informationsebene oberhalb des Genoms, die auch von den Umwelteinflüssen abhängt. Alterationen dieser Modifizierungen sind eines der Kernmerkmale der Krebsentstehung.
Ivosidenib ist als erster oraler Inhibitor des mutierten IDH-1 assoziiert mit einem günstigen Sicherheitsprofil (Durchfall 37%, Übelkeit, Obstipation, Hypokalzämie, Anämie 18,5%, Hyperglykämie, Kopfschmerzen, Pruritus und Fatigue je 14,8%).17 Schon initial konnte gezeigt werden, dass die Ivosidenib-Gabe prä- und postoperativ eine bessere Wirksamkeit bei nicht Kontrastmittel-aufnehmenden progredienten oder rezidivierten Grad-2/3-Gliomen zeigt und diese Substanzklasse eine Krankheitsstabilisierung und Verlangsamung der Wachstumsrate in den Vordergrund rückt. Zur Verlaufsbeobachtung eignen sich primär die 3-D-volumetrischen Auswertungen.18 Vorasidenib ist ein weiterer potenter Inhibitor des mutierten IDH-1/2 mit einer sehr guten ZNS-Gängigkeit. In orthotopen Gliommodellen haben sowohl Ivosidenib 500mg 1x1 wie auch Vorasidenib 50mg 1x1 p.o. eine Reduktion der 2-Hydroxyglutarat- Konzentrationen um 92% bewirken können. Die Phase-I-Studie wurde im Februar 2023 in „Nature Medicine“ veröffentlicht.19 Fast zeitgleich wurden die positiven Ergebnisse der Phase-III-Studie zu den genannten Medikamenten veröffentlicht, sie werden demnächst zu einer Änderung der derzeit noch aktuellen Behandlungskonzepte führen.
Neues Konzept bei Rezidiv-Glioblastom
Beim Rezidiv-Glioblastom gibt es keine klaren Standards: Reoperation, Rebestrahlung, Temozolomid-Rechallenge, Lomustin, Bevacizumab oder in Abhängigkeit von den molekularen Markern (BRAF-V600-E- aktivierende Mutation oder NTRK-Fusion)20 eine pharmakologische BRAF-Hemmung mit Dabrafenib und Trametinib in Analogie zur ROAR-Studie21 können angeboten werden. Viele Fusionen resultieren in einer Aktivierung der Rezeptor-Tyrosinkinasen und bieten sich als Target für eine zielgerichtete Therapie an. Genfusionen stehen hinter der Entstehung progredienter Tumoren in bis zu 16,5% der Fälle, Genfusionen bei GB sind aber mit ca 1% sehr selten.22 Multiple Fusionen können auch beim Glioblastom gefunden werden (FGFR3-TACC3-, EGFR-, PDGFRA-, NTRK-1, ROS1- oder MET-, ALK-Fusionen).23 Auch NTRK-Inhibitoren (Larotrectinib und Entrectinib) können als individueller Heilversuch, wenn auch mit weniger Evidenz für einen klinischen Nutzen untermauert, eingesetzt werden. Der Einsatz molekular zielgerichteter Medikamente mit Zulassung in anderen Tumorbereichen und dokumentierter Wirksamkeit, gemessen an der ESMO-MCBS***,24 kann bei Gliomen im Sinne eines freien Therapieversuchs gemäß der DGN-Leitlinie erwogen werden.25
Regorafenib, ein Multikinase-Inhibitor, zeigte in einer randomisierten Phase-II-Studie (REGOMA) bei Patienten mit erstem Progress eines GB im Vergleich zu CCNU/Lomustin eine Verlängerung des progressionsfreien und des Gesamtüberlebens.26 Die Interimsanalyse des Regorafenib-Armes der Studie GB AGILE (NCT03970447) konnte diese Daten nicht bestätigen, was in einer Presseaussendung am 21.9.22 mitgeteilt wurde. Die endgültigen Daten werden noch erwartet.
Zuletzt sollen auch die Supportivtherapien richtig eingesetzt und sorgfältig überwacht werden.27
Behandlung neuer Tumortypender IDH-Wildtyp-Gliome
Das diffuse Mittelliniengliom, H3-K27-alteriert, wird gemäß der Leitlinie mit Radiotherapie (54–60 Gy in 1,8–2-Gy-Fraktionen) und evtl. Temozolomid behandelt. Aufgrund der sehr schlechten Prognose wird die Behandlung in klinischen Studien mit „small molecules“ wie ONC 201,28 einem Dopamin-Rezeptor-D2-Antagonisten und Agonisten der caseinolytischen Protease, ermutigt. Für diffuse hemisphärische Gliome, H3.3-G34-mutiert, wird initial eine kombinierte Radiochemotherapie empfohlen. Bei Progress werden beide Entitäten in Analogie zum GB behandelt. Mehr als je zuvor war in den letzten Jahren in der interdisziplinären Behandlung Flexibilität gefordert.
Die Entscheidung für eine tumorspezifische Therapie soll nach Integration aller neuen Erkenntnisse während der gesamten Behandlungstrajektorie immer interdisziplinär getroffen werden, und der Patientenwille hat die höchste Priorität (DGN-Leitlinie 2021).
Abkürzungen:
* c-IMPACT-NOW: Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy-Not Officially WHO
** MGMT: O-6-Methylguanin-DNA-Methyltransferase
*** ESMO (European Society for Medical Oncology) Magnitude of Clinical Benefit Scale, Form 3
Literatur:
1 Louis DN et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 2021; 23(8): 1231-51 2 Weller M et al.: EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol 2021; 18(3): 170-86 3 Wick W: Gliome, S2k-Leitlinie, 2021, in: Deutsche Gesellschaft für Neurologie (Hrsg.): Leitlinien für Diagnostik und Therapie in der Neurologie. Available from: www.dgn.org/leitlinien abgerufen am 13.03.2023 4 Brat DJ et al.: Molecular biomarker testing for the diagnosis of diffuse gliomas. Arch Pathol Lab Med 2022; 146(5): 547-74 5 Reinhardt A et al.: Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 2018; 136(2): 273-91 6 Capper D et al.: DNA methylation-based classification of central nervous system tumours. Nature 2018; 555(7697): 469-74 7 Bagley S et al.: Glioblastoma Clinical Trials: Current landscape and opportunities for improvement. Clin Cancer Res 2022; 28(4): 594-602 8 Rahman R et al.: Current drug development and trial designs in neuro-oncology: report from the first American Society of Clinical Oncology and Society for Neuro-Oncology Clinical Trials Conference. Lancet Oncol 2023; 24(4): e161-71 9 Shirahata M et al.: Novel, improved grading system(S) for IDH-mutant astrocytic gliomas. Acta Neuropathol 2018; 136(1): 153-66 10 Appay R et al.: CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol 2019; 21(12): 159-28 11 Stupp R et al.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352(10): 987-96 12 Herrlinger U et al.: Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA–09): a randomised, open-label, phase 3 trial. Lancet 2019; 393(10172): 678-88 13 Stupp R et al.: Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 2017; 318(23): 2306-16 14 Perry JR et al.: Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med 2017; 376(11): 1027-37 15 Van Den Bent MJ et al.: Second interim and first molecular analysis of the EORTC randomized phase III intergroup CATNON trial on concurrent and adjuvant temozolomide in anaplastic glioma without 1p/19q codeletion. J Clin Oncol 2019; 37(15_suppl): 2000 16 Lassman AB et al.: Joint Final Report of EORTC 26951 and RTOG 9402: Phase III trials with procarbazine, lomustine, and vincristine chemotherapy for anaplastic oligodendroglial tumors. J Clin Oncol 2022; 40(23): 2539-45 17 Mellinghoff I et al.: ACTR-66. A phase 1, open-label, perioperative study of ivosidenib (AG-120) and vorasidenib (AG-881) in recurrent IDH1 mutant, low-grade glioma: updated results. Neuro Oncol 2019; 21 (Supplement_6): vi28-9 18 Ellingson BM et al.: Volumetric measurements are preferred in the evaluation of mutant IDH inhibition in non-enhancing diffuse gliomas: evidence from a phase I trial of ivosidenib. Neuro Oncol 2022; 24(5): 770-8 19 Mellinghoff IK et al.: Vorasidenib and ivosidenib in IDH1-mutant low-grade glioma: a randomized, perioperative phase 1 trial. Nat Med 2023; 29(3): 615-22 20 Wen PY et al.: Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol 2022; 23(1): 53-64 21 Vivek S et al.: ROAR: a phase 2, open-label study in patients (PTS) with BRAF V600E-mutated rare cancers to investigate the efficacy and safety of dabrafenib (D) and trametinib (T) combination therapy. J Clin Oncol 2016; 34 (15) 22 Gao Q et al.: Driver fusions and their implications in the development and treatment of human cancers. Cell Rep 2018; 23(1): 227-38 23 You G et al.: Fusion genes altered in adult malignant gliomas. Front Neurol 2021; 12: 715206 24 Cherny NI et al.: ESMO-Magnitude of Clinical Benefit Scale version 1.1. Ann Oncol 2017; 28(10): 2340-66 25 Fougner V et al.: Implementing targeted therapies in the treatment of glioblastoma: previous shortcomings, future promises, and a multimodal strategy recommendation. Neurooncol Adv 2022; 4(1): vdac157 26 Lombardi G et al.: Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol 2019; 20(1): 110-9 27 Roth P et al.: Neurological and vascular complications of primary and secondary brain tumours: EANO-ESMO Clinical Practice Guidelines for prophylaxis, diagnosis, treatment and follow-up. Ann Oncol 2021; 32(2): 171-82 28 Wierzbicki K et al.: Targeting and therapeutic monitoring of H3K27M-mutant glioma. Curr Oncol Rep 2020; 22(2): 19. doi: 10.1007/s11912-020-0877-0
Das könnte Sie auch interessieren:

Pharmakologische und neuromodulatorische Behandlungen des Clusterkopfschmerzes
Die Behandlung des chronischen Clusterkopfschmerzes steht noch heute vor großen Herausforderungen. Am 19. European Headache Congress (EHC) 2025 präsentierte Dr. Anja Petersen, ...

Ist die ketogene Diät eine Präzisionsmedizin?
Die ketogenen Ernährungstherapien sind etablierte Behandlungsformen bei Epilepsie. Während sie primär bei therapierefraktären pädiatrischen Epilepsien eingesetzt werden, finden sie ...

Neues aus der Alzheimer’s Disease Drug Development Pipeline
Mit der weltweiten Zulassung der Amyloidantikörper Lecanemab und Donanemab ist erstmals eine kausale Behandlung der Alzheimerkrankheit möglich geworden. Die Behandlung setzt an der ...