
Molekulare und zelluläre Veränderungen in der Pathophysiologie der Herzinsuffizienz
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Herzinsuffizienz (HI) entsteht durch strukturelle und funktionelle Veränderungen des Herzens, die zu einer unzureichenden Pumpleistung führen. Zentral sind maladaptive Prozesse in neurohormoneller Aktivierung sowie im kardialen Remodeling und zelluläre Dysfunktionen, die einen Teufelskreis aus Progression und Symptomen wie Dyspnoe und Ödemen auslösen. Ein tieferes Verständnis dieser Mechanismen eröffnet neue therapeutische Ansätze und macht die Pathophysiologie zu einem spannenden Forschungsfeld.
Keypoints
-
HFpEF wird zunehmend als Multisystemsyndrom mit extrakardialer Beteiligung gesehen, das primär durch systemische Inflammation, mikrovaskuläre Dysfunktion undmetabolischen Stress getriggert wird.
-
Die Pathophysiologie der HFrEF ist stärker kardiozentrisch und durch den Verlust funktionsfähiger Kardiomyozyten, neurohormonelle Aktivierung und maladaptives Remodeling gekennzeichnet.
-
Bei HFpEF stehen das metabolische Remodeling und die Immunaktivierung im Vordergrund.
-
Bei der HFrEF ist eher die mitochondriale Dysfunktion, die enger mit Myozytenverlust und neurohormonellem Stress assoziiert ist, zentral.
-
In HFrEF-Kardiomyozyten ist die Ca2+-Freisetzung vermindert, bei HFpEF jedoch gleichbleibend oder gesteigert.
Herzinsuffizienz betrifft weltweit über 64 Millionen Menschen und weist in wirtschaftlich entwickelten Ländern eine Prävalenz von etwa 1–3% bei Erwachsenen auf. Sie wird als die Unfähigkeit des Herzens definiert, eine ausreichende Blutmenge zu fördern, um den metabolischen Bedarf des Körpers zu decken. HI wird als ein komplexes klinisches Syndrom, das durch typische Symptome wie Dyspnoe, Müdigkeit und periphere Ödeme gekennzeichnet ist, beschrieben. Diese können sowohl unter Belastung als auch in Ruhe auftreten. Die Ursachen sind multifaktoriell und umfassen strukturelle oder funktionelle kardiale Störungen, die zu einer verminderten kardialen Auswurfleistung oder erhöhten Füllungsdrücken führen. Die Symptomatik bringt in der Regel eine signifikanten Einschränkung der Lebensqualität der Betroffenen mit sich.1
Klassifikation der Herzinsuffizienz: HFrEF vs. HFmrEF vs. HFpEF
Die echokardiografische Einteilung der HI erfolgt anhand der linksventrikulären Ejektionsfraktion (LVEF) und umfasst drei Hauptkategorien: 1) Herzinsuffizienz mit reduzierter LVEF (HFrEF) bei einer LVEF von ≤40%; 2) Herzinsuffizienz mit leicht reduzierter LVEF (HFmrEF) bei einer LVEF von 41–49% und 3) Herzinsuffizienz mit erhaltener LVEF (HFpEF) bei einer LVEF von ≥50%. In der Bestimmung der EF bestehen jedoch methodenabhängige Unterschiede, etwa zwischen Echokardiografie und Magnetresonanztomografie, die zu abweichenden Ergebnissen führen können.
Diese Kategorien unterscheiden sich nicht nur hinsichtlich der Auswurffraktion, sondern auch in ihren pathophysiologischen Grundlagen.
Das American College of Cardiology, die American Heart Association und die Heart Failure Society of America betonen, dass die Einteilung der Herzinsuffizienz nach LVEF sowohl prognostische als auch therapeutische Konsequenzen hat und die Wirksamkeit bestimmter Therapien mit steigender LVEF abnimmt. Während einige Therapien in allen Kategorien Vorteile zeigen, nimmt der Nutzen insbesondere bei LVEF ≥55–60% deutlich ab, was vermutlich auf die unterschiedlichen pathophysiologischen Mechanismen zurückzuführen ist.2
Auslöser und kardiale Veränderungen in der Pathophysiologie der Herzinsuffizienz
Die Pathophysiologie der Herzinsuffizienz ist durch eine Kaskade von Prozessen gekennzeichnet, die durch ein auslösendes Ereignis oder Komorbiditäten initiiert werden. Bei der HFrEF stehen meist ischämische Herzerkrankungen, Myokardinfarkt, Myokarditis, dilatative Kardiomyopathie oder Klappenerkrankungen im Vordergrund. Im Gegensatz dazu entsteht die HFpEF überwiegend durch Komorbiditäten wie Druckbelastung (z.B. arterielle Hypertonie), Volumenbelastung und weitere Faktoren. Das initiale kardiale Ereignis führt zu einer verminderten Herzleistung, woraufhin kompensatorische Mechanismen aktiviert werden, insbesondere die neurohormonelle Stimulation des sympathischen Nervensystems sowie des Renin-Angiotensin-Aldosteron-Systems (RAAS). Diese Mechanismen sind anfangs kompensatorisch, führen jedoch im Verlauf zu strukturellen Veränderungen des Myokards sowie Kontraktilitätsstörungen und begünstigen die Entstehung eines arrhythmogenen Substrats. In fortgeschrittenen Stadien treten zudem kardiorenale und kardiopulmonale Syndrome auf.3,4
Auf zellulärer und molekularer Ebene ist Herzinsuffizienz durch eine Hypertrophie der Kardiomyozyten sowie durch Veränderungen der extrazellulären Matrix gekennzeichnet. Zusätzlich sind Entzündungsprozesse, Fibrose, endotheliale Dysfunktion, Störungen der Sarkomerfunktion, mitochondriale und metabolische Defekte sowie Veränderungen in der NO-cGMP-PKG-Signalkaskade (engl. „nitric oxide-cyclic guanosine monophosphate-protein kinase G“) zentrale pathophysiologische Merkmale der Herzinsuffizienz. Diese Umbauprozesse führen zu strukturellen und funktionellen Veränderungen des Myokards, die eine Kontraktilitätsstörung und die Entstehung eines arrhythmogenen Substrats begünstigen. Auch Apoptose und andere Formen des programmierten Zelltods tragen wesentlich zum Verlust kontraktiler Kardiomyozyten und zur Entwicklung der ventrikulären Dysfunktion bei.
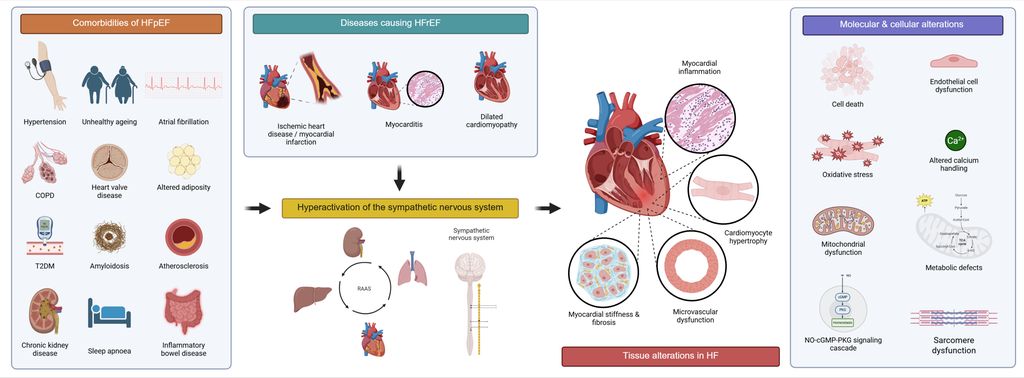
Abb. 1: Grafische Darstellung der Pathophysiologie von Herzinsuffizienz, die mit molekularen und zellulären Veränderungen des Herzens assoziiert ist
Kardiales Remodeling: molekulare Mechanismen & Mediatoren
Systemische Inflammation ist ein zentrales Merkmal der Herzinsuffizienz, tritt jedoch bei der HFpEF besonders deutlich hervor, während bei der HFrEF die neurohormonelle Aktivierung stärker ausgeprägt ist. Bei HFpEF wird die Entzündungsreaktion vor allem durch die häufig vorhandenen Komorbiditäten ausgelöst und zeigt sich in einer erhöhten Expression und Sekretion proinflammatorischer Zytokine wie Tumornekrosefaktor (TNF), TNF-Rezeptoren, Interleukin6 (IL-6) und Myeloperoxidase (MPO). Zusätzlich sind vermehrt Adhäsionsmoleküle wie das vaskuläre Zelladhäsionsmolekül 1 (VCAM-1) im Blut nachweisbar, was die Migration von Immunzellen (z.B. Monozyten, Makrophagen, dendritische Zellen) ins Myokard fördert und ein proinflammatorisches Milieu schafft. Diese kardiale Entzündung begünstigt pathologische Umbauprozesse wie die Myokardfibrose, die zu einer Verschlechterung der myokardialen Funktion führt. Darüber hinaus tragen die freigesetzten Entzündungsmediatoren zur Endothelfunktionsstörung und zu vermehrtem oxidativem Stress bei, wodurch ein Teufelskreis der Schädigung entsteht.3
Kardiale Fibrose ist ein zentrales Merkmal der Herzinsuffizienz. Multi-Omics-Analysen haben gezeigt, dass verschiedene Fibroblastensubpopulationen mit jeweils spezifischen transkriptionellen und epigenetischen Profilen an der Entstehung der kardialen Fibrose beteiligt sind. Diese Subpopulationen exprimieren Marker wie Periostin (POSTN), Fibroblast Activation Protein (FAP) und Adipocyte Enhancer Binding Protein 1 (AEBP1), die mit ungünstigen Gewebeumbauprozessen assoziiert sind und als zirkulierende Biomarker für den Schweregrad und die Progression der Erkrankung dienen können. Darüber hinaus wurden erhöhte Spiegel weiterer zirkulierender Biomarker wie Fibroblast Growth Factor 23 (FGF23), Chitinase-3-like Protein 1 (CHI3L1), Suppression of Tumorigenicity 2 (ST2) und Matrix-Metalloproteinase 7 (MMP7) sowie eine Reduktion von Matrix-Metalloproteinase1 (MMP1) als prognostische Marker, insbesondere bei HFpEF, identifiziert. Pathophysiologisch entsteht Fibrose durch die Interaktion von Immunzellen (insbesondere CCR2-positiven Makrophagen), Fibroblasten und inflammatorischen Zytokinen wie Interleukin1β (IL-1β) oder durch die Aktivierung des Transforming-Growth-Factor-Beta(TGF-β)-Signalwegs. Die Hemmung des IL-1β-Signalwegs stellt einen vielversprechenden therapeutischen Ansatz dar, der in präklinischen Modellen bereits zu einer Reduktion der kardialen Fibrose und einer Verbesserung der Herzfunktion geführt hat. Aufgrund der vielfältigen Funktionen von TGF-β konzentrieren sich potenzielle therapeutische Strategien eher auf die Modulation von Regulatoren stromauf- oder stromabwärts des Signalwegs.2,4,5
Aktuelle Forschungsergebnisse unterstreichen die zentrale Bedeutung der endothelialen Dysfunktion, die mittlerweile als bidirektionaler Faktor sowohl bei der HI mit reduzierter als auch mit erhaltener Ejektionsfraktion gilt. Charakteristisch für die Endothelstörung sind eine verminderte Verfügbarkeit von Stickstoffmonoxid, vermehrter oxidativer Stress sowie ein proinflammatorischer und prothrombotischer Zustand. Dies führt zu gestörter Vasodilatation sowie erhöhter Gefäßsteifigkeit und fördert kardiovaskuläres Remodeling. Bei der HFrEF sind insbesondere die neurohormonelle Aktivierung und die gesteigerte Bildung reaktiver Sauerstoffspezies für die Endothelschädigung verantwortlich. Im Gegensatz dazu begünstigen bei der HFpEF Komorbiditäten wie Adipositas, Diabetes mellitus und Hypertonie einen systemischen Entzündungszustand, der zu einer Entzündung des mikrovaskulären Endothels führt. Dies triggert wiederum Myokardfibrose und eine erhöhte Steifigkeit des Herzmuskels. Zahlreiche Studien zeigen zudem, dass eine Endothelfunktionseinschränkung mit ungünstigeren klinischen Verläufen assoziiert ist.6
Mitochondriale und metabolische Störungen gelten heute als Schlüsselfaktoren in der Entstehung und dem Fortschreiten der HI. Sowohl bei der HI mit reduzierter als auch mit erhaltener Ejektionsfraktion sind sie eng mit der Entwicklung einer kardialen Fibrose verbunden. Eine gestörte mitochondriale Funktion beeinträchtigt die oxidative Phosphorylierung, was zu einer Reduktion der ATP-Reserven führt und die Energieversorgung der Kardiomyozyten limitiert. Dies begünstigt die Schädigung und Apoptose der Herzmuskelzellen. Insbesondere bei HFpEF in Kombination mit metabolischem Syndrom oder Diabetes kommt es zu einer Überlastung der Mitochondrien durch ein Überangebot an Substraten. Dies führt zur Akkumulation toxischer Metaboliten, zu vermehrter Bildung reaktiver Sauerstoffspezies (ROS) und Störungen im Ionenhaushalt, insbesondere des Kalziums (Ca2+). Die daraus resultierenden energetischen Defizite und der oxidative Stress aktivieren profibrotische Signalwege, wie den TGF-β, sowie inflammatorische Kaskaden, was die kardiale Fibrose und das Fortschreiten der HI weiter fördert.4,7
Aktuelle Forschungsergebnisse belegen, dass der Stickstoffmonoxid-cGMP-Proteinkinase-G(NO-cGMP-PKG)-Signalweg eine zentrale Rolle bei der Regulation des kardialen Remodelings sowie der mitochondrialen und metabolischen Homöostase spielt. Insbesondere bei HFpEF ist dieser Signalweg supprimiert, was zu einer verminderten Phosphorylierung essenzieller Zielproteine führt, die für die mitochondriale Biogenese und Funktion relevant sind. Zudem resultiert die Hypophosphorylierung von Titin in Kardiomyozyten in einer erhöhten Steifigkeit der Herzmuskelzellen. Im Kontext der kardialen Fibrose fördert eine reduzierte NO-cGMP-PKG-Aktivität die Aktivierung von Fibroblasten und die vermehrte Ablagerung von Kollagen in der extrazellulären Matrix. Vergleichbare Veränderungen werden auch bei HFrEF beobachtet, wobei die zugrundeliegenden Auslöser unterschiedlich sind.2,8
Die Dysfunktion der Sarkomere ist ein zentrales Element der HI, wobei sich die zugrundeliegenden Mechanismen und Folgen zwischen HFpEF und HFrEF unterscheiden. Bei HFpEF steht vor allem eine erhöhte passive Steifigkeit des sarkomerischen Proteins Titin im Vordergrund. Diese resultiert aus einer Hypophosphorylierung von Titin, was die Relaxation der Kardiomyozyten beeinträchtigt und zur diastolischen Dysfunktion führt. Im Gegensatz dazu ist die sarkomerische Dysfunktion bei HFrEF vor allem durch den Verlust kontraktiler Kardiomyozyten gekennzeichnet. Hier liegen Veränderungen in der Expression und posttranslationalen Modifikation sarkomerischer Proteine vor, was zu einer verminderten systolischen Leistungsfähigkeit führt. Zusammengefasst resultiert die diastolische Dysfunktion bei HFpEF primär aus einer erhöhten Steifigkeit der Sarkomere, während bei HFrEF der systolische Funktionsverlust durch strukturelle und funktionelle Veränderungen der Sarkomere dominiert.9
Zentrales pathophysiologisches Merkmal: Störungen der Kalziumhomöostase
Der enge Zusammenhang zwischen der kardialen Kontraktion und den zugrundeliegenden Ionenströmen ist bereits in frühen Arbeiten umfassend beschrieben worden und gilt als gesichert. Insbesondere Ca2+ übernimmt hierbei eine Schlüsselrolle. Störungen in der Ca2+-Regulation beeinflussen die Erregungs- und Kontraktionskopplung der Kardiomyozyten maßgeblich. Die Mechanismen der Ca2+-Dysregulation unterscheiden sich jedoch deutlich zwischen HFpEF und HFrEF: Während bei HFrEF eine gestörte Ca2+-Freisetzung und -Aufnahme mit verminderter Kontraktilität und gestörter Relaxation im Vordergrund steht, zeigen HFpEF-Modelle eine verstärkte Kopplungsfidelität, aber eine diastolische Ca2+-Überladung und Steifigkeit der Herzmuskelzellen, was zu unterschiedlichen pathophysiologischen Konsequenzen führt (Abb.2).4,9,10
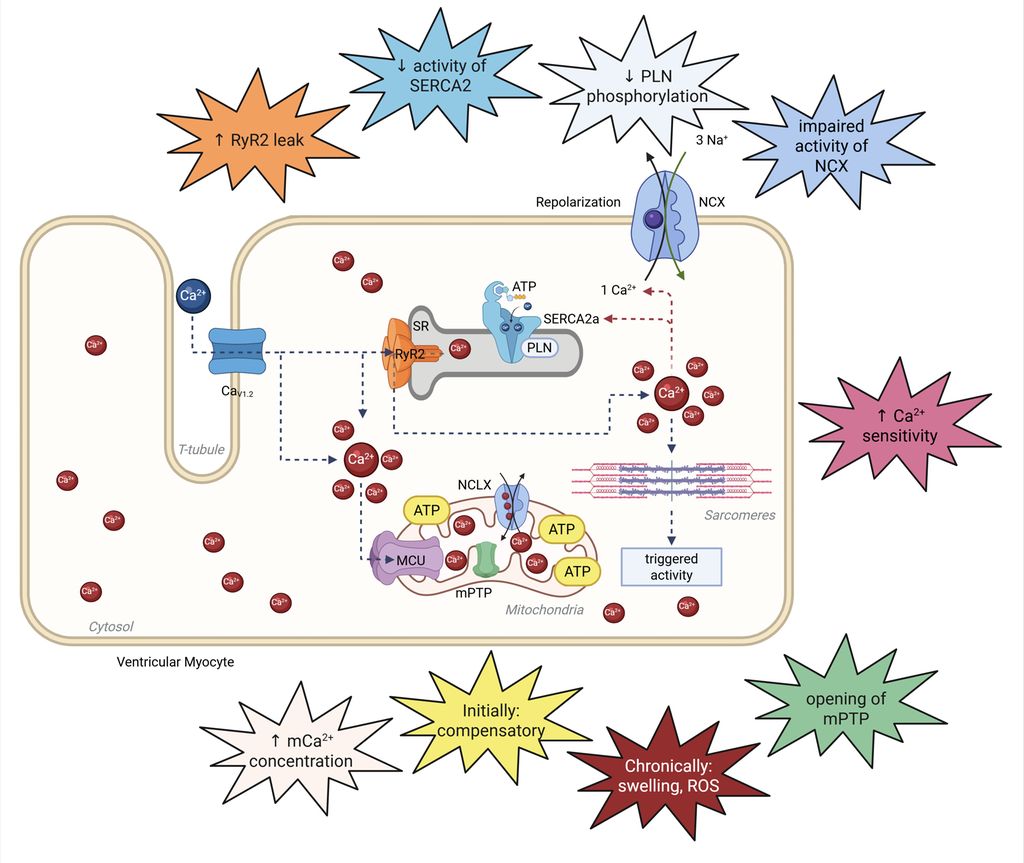
Abb. 2: Herzinsuffizienz führt zu Veränderungen in der Kalziumhomöostase. Durch Fehlregulationen der Kalziumströme kommt es zu den bei der Herzinsuffizienz typischen systolischen bzw. diastolischen Dysfunktionen
Die HFrEF ist durch eine gestörte Erregungs- und Kontraktionskopplung gekennzeichnet, wobei insbesondere eine verminderte Dichte und Desorganisation der T-Tubuli zu einer abgeschwächten und asynchronen Ca2+-Freisetzung führen. Dies resultiert in reduzierten Ca2+-Ionen und beeinträchtigter Kontraktilität der Kardiomyozyten. Zusätzlich sind die Expression und die Funktion des L-Typ-Ca2+-Kanals (LTCC) herabgesetzt, was die Ca2+-Einstromdynamik weiter verschlechtert.11 Bei HFrEF kann die Wiederaufnahme von Ca2+ in das sarkoplasmatische Retikulum (SR) durch eine reduzierte Aktivität der SR-Kalzium-ATPase2a (SERCA2a) und eineHypophosphorylierung von Phospholamban eingeschränkt sein, was die diastolische Ca2+-Clearance limitiert. Dies ist allerdings nicht die primäre Abnormalität der HFrEF. Gleichzeitig ist der Ryanodinrezeptor2 (RyR2) am SR häufig hyperphosphoryliert und oxidiert, was zu einer erhöhten, unkontrollierten Ca2+-Freisetzung („leaky RyR2“) und damit zu einem Ca2+-Verlust aus dem SR und folglich zur erhöhten Arrhythmieanfälligkeit führt. Die Aktivität des Natrium-Ca2+-Austauschers (NCX) ist bei HFrEF oft gesteigert, wodurch vermehrt Ca2+ aus der Zelle entfernt wird und das SR-Ca2+-Reservoir weiter abnimmt. Dies trägt zu einer weiteren Schwächung der systolischen und diastolischen Funktion sowie zur Arrhythmogenese bei. Darüber hinaus ist die mitochondriale Funktion bei HFrEF beeinträchtigt: Die Ca2+-Aufnahme in die Mitochondrien ist reduziert, was die ATP-Produktion limitiert und oxidativen Stress fördert. Insgesamt resultieren diese Veränderungen in einer Sarkomerdysfunktion, verminderter Kontraktilität und maladaptivem kardialem Remodeling, einschließlich Fibrose.10
Im Gegensatz zur HFrEF zeigt die HFpEF häufig eine unveränderte oder sogar gesteigerte systolische Ca2+-Amplitude. Hingegen zeichnet sich die HFpEF durch eine obligate starke Beeinträchtigung der diastolischen Ca2+-Entfernung aus, was zu einer erhöhten diastolischen Ca2+-Konzentration in den Kardiomyozyten führt und eine unvollständige Relaxation der Herzmuskelzellen verursacht. Zudem ist ebenso das Risiko einer unkontrollierten Ca2+-Freisetzung durch einen „leaky RyR2“ erhöht. In den Mitochondrien finden sich pathologische Veränderungen Ca2+-assoziierter Proteine und eine erhöhte mitochondriale Ca2+-Konzentration, was zytotoxisch wirkt, die respiratorische Kapazität mindert und die metabolische Flexibilität der Zellen einschränkt. Die T-Tubulus-Struktur bleibt bei HFpEF in der Regel erhalten oder ist sogar verstärkt, sodass die Erregungs- und Kontraktionskopplung effizient bleibt, während die diastolische Dysfunktion im Vordergrund steht.12
Die beschriebene Hypophosphorylierung von Titin führt in beiden Syndromen zu einer Zunahme der passiven Steifigkeit der Kardiomyozyten. Dies stellt eine direkte Verbindung zwischen der gestörten Ca2+-Ionen-Regulation, Veränderungen auf Sarkomerebene und der diastolischen Dysfunktion her.9,10
Fazit
Abschließend lässt sich festhalten, dass die Pathophysiologie der Herzinsuffizienz durch ein komplexes Zusammenspiel von strukturellen, neurohormonellen, zellulären und molekularen Mechanismen gekennzeichnet ist, deren Verständnis eine essenzielle Grundlage für die Entwicklung gezielter diagnostischer und therapeutischer Strategien bildet.
Literatur:
1 Khan MS et al.: Global epidemiology of heart failure. Nat Rev Cardiol 2024; 21(10): 717-34 2 Hamo CE et al.: Heart failure with preserved ejection fraction. Nat Rev Dis Primers 2024; 10(1):55 3 Alcaide P et al.: Myocardial inflammation in heart failure with reduced and preserved ejection fraction. Circ Res 2024; 134(12): 1752-66 4 Méndez-Fernández A et al.: Distinguishing pathophysiological features of heart failure with reduced and preserved ejection fraction: a comparative analysis of two mouse models. J Physiol 2024: doi: 10.1113/JP286410 5 Ghazal R et al.: Cardiac fibrosis in the multi-omics era: implications for heart failure. Circ Res 2025; 136(7):773-802 6 Drera A et al.: Endothelial dysfunction in heart failure: what is its role? J Clin Med 2024; 13(9): 2534 7 Gallo G et al.: Mitochondrial dysfunction in heart failure: from pathophysiological mechanisms to therapeutic opportunities. Int J Mol Sci 2024; 25(5): 2667 8 Cai Z et al.: The NO-cGMP-PKG axis in HFpEF: from pathological mechanisms to potential therapies. Aging Dis 2023; 14(1): 46-62 9 Aksentijevic D et al.: Mechano-energetic uncoupling in heart failure. Nat Rev Cardiol 2025: doi: 10.1038/s41569-025-01167-6 10 Liu YB et al.: Abnormal phosphorylation / dephosphorylation and Ca(2+) dysfunction in heart failure. Heart Fail Rev 2024; 29(4): 751-68 11 Khalid A et al.: Molecular mechanisms of L-type calcium channel dysregulation in heart failure. Int J Mol Sci 2025; 26(12): 5738 12 Rosas PC et al.: Early pathological mechanisms in a mouse model of heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol 2024; 327(6): 1524-43
Das könnte Sie auch interessieren:
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...
Antibiotika bei Endokarditis: ambulante Fortsetzung der stationären Therapie
Nach einer erfolgreichen initialen stationären, parenteralen Antibiotikatherapie besteht bei vielen Patient:innen mit bakterieller Endokarditis die Option einer ambulanten ...