How to treat CTEPH im Jahr 2022?
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die chronisch thromboembolische pulmonale Hypertension ist eine seltene Erkrankung, die durch eine ungenügende Auflösung von Thromboembolien in den Lungenarterien entsteht. Große Serien konnten die Entstehung einer CTEPH nach akuter PE nur bei einem kleinen Prozentsatz der Patienten nachweisen. Daher gib es keinerlei Prophylaxe für CTEPH.
Keypoints
-
Die Therapie der Wahl bei CTEPH ist die pulmonale Endarterektomie (PEA), die eine 30-Tages-Mortalität von 5% aufweist.
-
In 17–31% der Fälle kommt es nach einer PEA zu keiner ausreichenden Lungenhochdrucksenkung.
-
Als Alternative für diese Patienten steht die Ballonangioplastie (BPA) der Lungenarterien zu Verfügung.
-
Registerdaten zeigen nach stattgehabter BPA eine symptomatische Verbesserung und eine 30-Tages-Mortalität <2%.
-
Die Kombination der BPA mit medikamentösen Therapien (Antikoagulation, Riociguat, Treprostinil) kann zu einer weiteren signifikanten Verbesserung für Patienten führen.
Obwohl die pulmonale Endarterektomie (PEA) die Therapie der Wahl darstellt, werden wenigstens die Hälfte aller CTEPH-Patienten nicht operiert oder leiden nach einer Operation weiterhin an Lungenhochdruck. Welche Patienten nach Operation einen unbefriedigenden Erfolg erreichen, ist präoperativ nicht mit Sicherheit zu sagen. Allerdings steht für diese Patienten ein neues interventionelles Therapieverfahren, die Ballonangioplastie (BPA) der Lungenarterien, zur Verfügung. Registerdaten zeigen eine hämodynamische und symptomatische Verbesserung mit einer 30-Tages-Mortalität <2%. Es zeichnet sich ab, dass die medikamentösen Behandlungen in Kombination mit BPA zu weiteren signifikanten Verbesserungen führen, daher wird häufig schon vor der Entscheidung zu einer mechanischen Therapie mit gefäßerweiternden Substanzen begonnen. Die drei Therapieoptionen ergänzen einander. In welcher Reihenfolge diese Komplementarität stattfindet, wird vom CTEPH-Team festgelegt.
Epidemiologie und Pathophysiologie
CTEPH ist eine relativ seltene, aber potenziell lebensbedrohliche Erkrankung der pulmonalen arteriellen Gefäße und ist Teil der Gruppe 4 der WHO-Klassifikation für pulmonale Hypertension.1 CTEPH ist die Folge von Thromboembolien der Lungenarterien, die sich nicht auflösen, sondern sich in fibröses Material umwandeln, was zu einem Verschluss der Pulmonalarterien führt, zu einem Remodelling der Mikrozirkulation und zu mehr pulmonaler Hypertension (Lungenhochdruck). Histologisch reichen die organisierten Gerinnsel bis zu einem Gefäßdurchmesser von 200µm. Die Inzidenz von CTEPH nach akuter Lungenembolie lag bei einer rezenten hochwertigen prospektiven Kohortenbeobachtungsstudie(FOKUS) kumulativ bei 2,3% nach 2 Jahren.2
Klinik und Diagnostik
Symptome der Rechtsherzerkrankung treten spät auf und sind nicht spezifisch. Es gibt auch keine verlässlichen CTEPH-spezifischen Symptome, die es erlauben, die Erkrankung von anderen Formen der pulmonalen Hypertension zu unterscheiden. Die Diagnostik gestaltet sich selbst an spezialisierten Zentren schwierig mit durchschnittlich 14 Monaten zwischen Symptombeginn und Diagnose. Nicht alle CTEPH-Patienten berichten in ihrer Vorgeschichte über eine akute Lungenembolie.
Die wichtigsten prädisponierenden Faktoren für eine CTEPH sind in aufsteigender Reihenfolge ventrikuloatriale Shunts oder Schrittmachersonden, der Zustand nach Splenektomie, große und rezidivierende venöse Thromboseereignisse, Hypothyreose mit Hormonersatz, entzündliche Darmerkrankungen und Karzinome.3
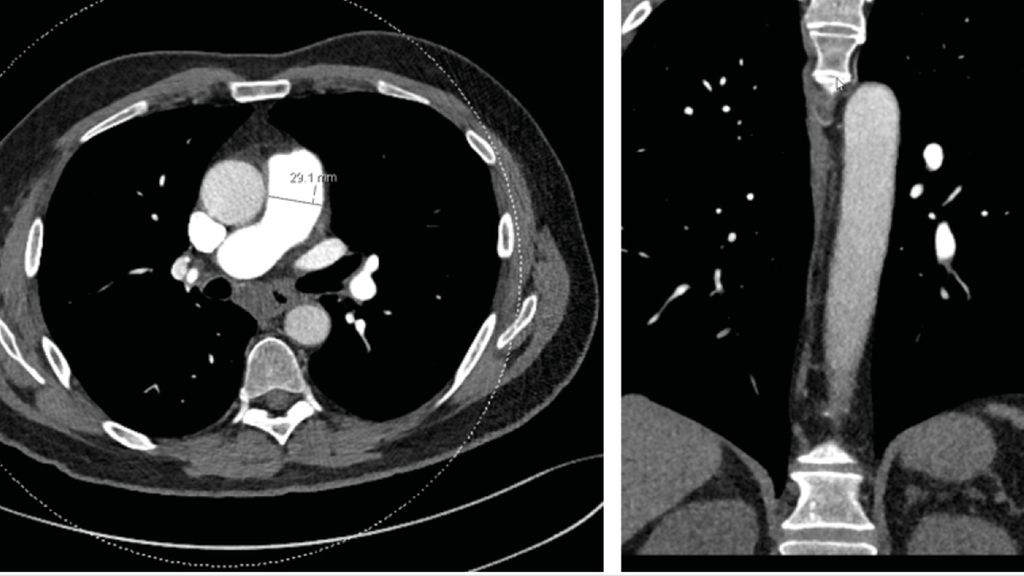
Die diagnostischen Kriterien umfassen eine invasiv bestätigte pulmonale Hypertension mit einem pulmonalen Mitteldruck (mPAP) von >20mmHg und einem Wedge-Druck ≤15mmHg, segmentale Perfusionsdefizite bei erhaltener Ventilation im V/Q-Szintigramm (Abb. 1C) sowie eine CT (Abb. 1A und 1B) oder invasive Angiografie mit den typischen vaskulären Läsionen einer CTEPH.1 Dazu gehören ringförmige Stenosen, Webs, hochgradige oder chronische Verschlüsse und Tortuositäten. Diese Veränderungen sind teilweise schwierig zu erkennen mittels CT oder der nicht selektiven Pulmonalisangiografie, weswegen diese Verfahren alleine nicht zum Ausschluss einer CTEPH genügen. Geprüft wird derzeit die Anwendung des „Dual Energy CT“ (Abb. 1D und 1E), welche sowohl Anatomie als auch Perfusion darstellt, und/oder die Anwendung der selektiven Angiografie einzelner Pulmonalarterien. Symptomatische Patienten nach akuter Lungenembolie mit einem Perfusionsdefizit im V/Q-Scan >3 Monate nach Pulmonalembolie sollten am PH-Zentrum abgeklärt werden. Das Screening für CTEPH bei asymptomatischen Risikopatienten ist gemäß den neusten ESC-Richtlinien eine Klasse-I-Empfehlung.1
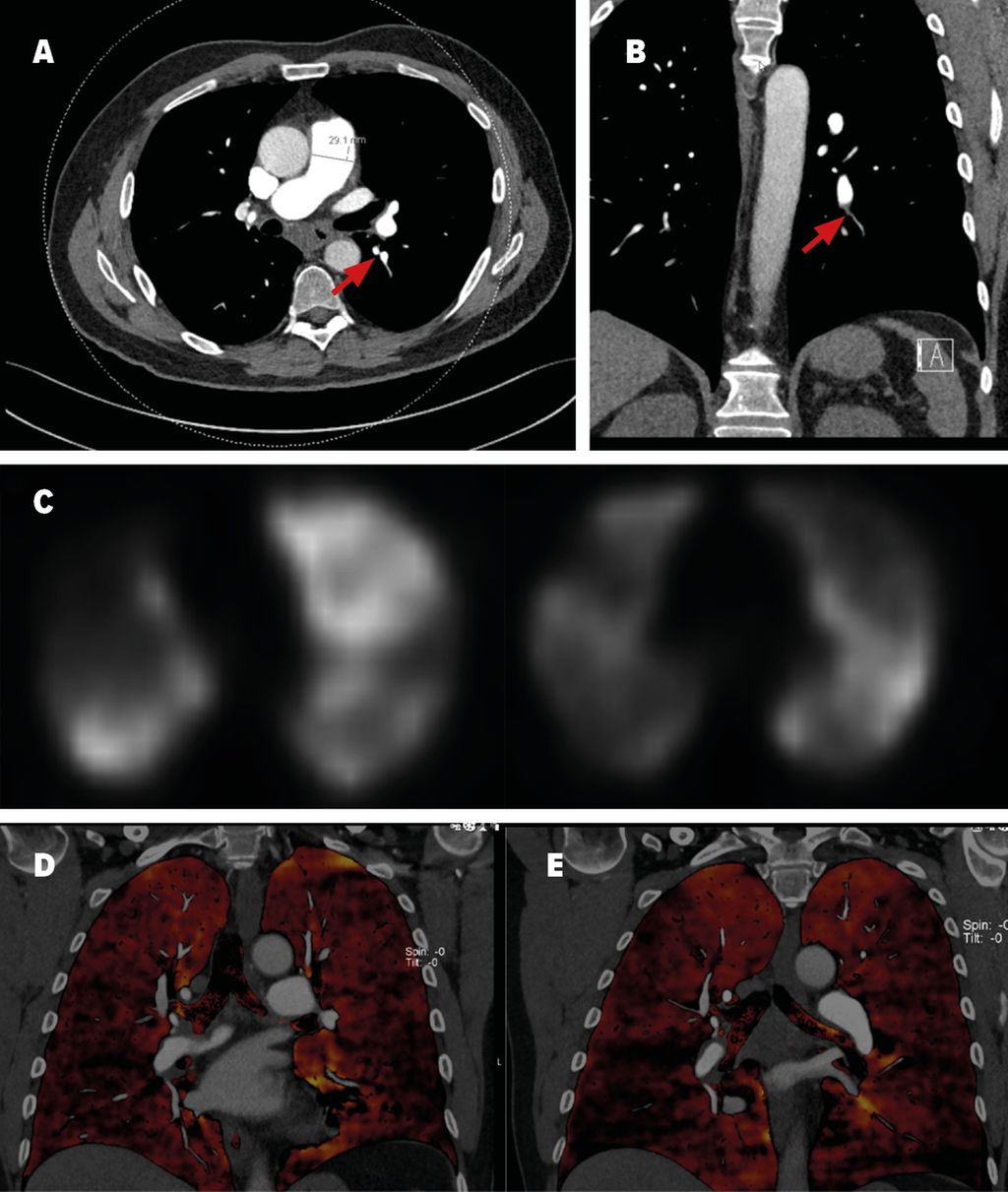
Abb. 1: Bildgebung eines CTEPH-Patienten: A und B: CT-Thorax mit Kontrastmittel mit indirekten Zeichen für eine pulmonale Hypertonie, wie grenzwertigem Diameter des Truncus pulmonalis von 29 mm und peripherer Gefäßrarefizierung. Begleitend liegt eine infrakarinäre Lymphadenopathie vor, am ehesten im Rahmen der pulmonalen Druckerhöhung. Die roten Pfeile markieren Kalibersprünge der Pulmonalarterien als Zeichen für intravaskuläres Material. C: Ventilations-/Perfusionsszintigrafie: multiple segmentale bis subsegmentale Minderanreicherung in der Perfusions-SPECT passend zur distalen Form der CTEPH. D und E: «Dual Energy»-CT-Angiografie der Lunge mit Visualisierung der Iodverteilung als Korrelat der Lungenperfusion (schwarze Areale entsprechen Perfusionsdefekten respektive Artefakten). C und D: In Abb. C Perfusionsdefizite (Unterlappen rechts und Lingula) vor Behandlung, während sich in Abb. D eine Befundbesserung nach BPA zeigt.
Pulmonale Endarterektomie und Ballonangioplastie
Die Therapie der Wahl bei symptomatischer CTEPH ist die pulmonale Endarterektomie. Der Eingriff bedingt eine tiefe Hypothermie mit Kreislaufstillstand. Die 30-Tages-Mortalität der PEA liegt bei ca. 5% und an sehr erfahrenen Zentren weniger als 2%. Die Operabilität sollte von einem interdisziplinären Team beurteilt werden: Die Thromben sollten der Chirurgie zugänglich sein, d.h. im Hauptstamm, lobär und segmental, und es sollten keine prohibitiven Komorbiditäten vorhanden sein und keine sonstigen Limitationen, die das Risiko-Nutzen-Verhältnis relevant einschränken (z.B. Gebrechlichkeit). Gemäß dem europäischen CTEPH-Register waren von 679 untersuchten Patienten 36,5% als nicht operabel eingestuft und von den operablen Patienten wiederum wurden 14% nie operiert. Mit der Operation wird meistens eine Normalisierung des mPAP und Symptomfreiheit erreicht, der Therapieansatz ist also durchaus kurativ. Bei 17–31% kommt es allerdings zu einer nicht ausreichenden Drucksenkung wegen inkompletter Entfernung der Obstruktionen, Reperfusionsödem oder schwerer sekundärer Mikrovaskulopathie. Wer nach Operation weiterhin Lungenhochdruck hat, kann präoperativ nicht sicher vorausgesagt werden. Zusammenfassend werden also fast 50% der CTEPH-Patienten keiner Operation unterzogen,4 und weitere 20% haben weiterhin Lungenhochdruck.
Als Alternative für diese Patienten hat sich in der letzten Dekade die Ballonangioplastie der Lungenarterien etabliert, für welche die aktuellen PH-Richtlinien eine Klasse-IA-Indikation vorsehen. Welche Patienten am besten von einer BPA profitieren, bleibt derzeit unklar. Die erste BPA wurde 1988 in Leiden, Holland, bei einem 30-Jährigen mit CTEPH erfolgreich durchgeführt mit einer mPAP- Senkung von 46 auf 35mmHg.5 Eine erste, 18 Patienten umfassende Serie wurde an der Harvard Medical School in Boston zwischen 1994 und 1999 behandelt.6 BPA war nach durchschnittlich nur 2,6 Interventionen in insgesamt 6 Segmenten erfolgreich (mPAP-Senkung von 43 auf 34mmHg), allerdings trat in 61% der Fälle ein Lungenschaden auf, der in 17% eine mechanische Ventilation bedingte. Die Mortalität war mit 5,7% hoch. Erst eine modifizierte Form der BPA, entwickelt in Okayama, Japan, hat der Methode neuen Aufwind gegeben und die aktuelle Entwicklung weltweit ermöglicht. Zusammengefasst lässt sich eine Lungengefäßwiderstandssenkung um 65% erreichen bei einer periprozeduralen Mortalität von durchschnittlich 1,8%. Während bei einem mPAP >40 nicht mehr als 2–3 Läsionen angegangen werden sollten, sind bei einem mPAP um 35mmHg und weniger nur die Kontrastmittelmenge, die Bestrahlungszeit und die Ermüdung des Patienten limitierend. Während der BPA können in ca. 9% der Sitzungen Lungenblutungen auftreten durch Drahtperforation der distalen Äste oder Perforation mit Ballonen. Bereits sehr kleine Blutungen, die angiografisch nicht zwingend erkennbar sind, führen zu Husten (mit oder ohne Hämoptyse). Ein konventionelles Lungenröntgen zum Ausschluss von Infiltraten wird routinemäßig durchgeführt. Die Entlassung kann in der Regel am Folgetag stattfinden. Das europäische BPA-Register wird mithelfen, offene Fragen zu beantworten.
Medikamentöse Behandlung
Die medikamentöse Therapie beinhaltet eine konventionelle orale Antikoagulation (OAK; Klasse-I-Indikation)3 und den Guanylatcyclase-Stimulator Riociguat für die Behandlung der inoperablen oder persistenten/rezidivierenden CTEPH.4 Für schwere PH mit Lungengefäßwiderständen >800dynes.cm.s–5 ist subkutanes Treprostinil anzuwenden und auch zugelassen.5
Die Frage, wie medikamentöse Therapien, zum Beispiel Riociguat im Vergleich zu BPA, bei inoperabler CTEPH wirken, wurde durch zwei rezente Studien beantwortet. Während der Effekt der BPA auf den Lungengefäßwiderstand größer ist als der durch Riociguat in Höchstdosis erreichbare Effekt, kommt es durch BPA zu einer deutlichen Reduktion des pulmonalarteriellen Mitteldrucks, während Riociguat vor allem das Herzzeitvolumen erhöht.
Fazit
Die modernen Therapien der CTEPH haben in einem multimodalen Zusammenwirken die Aussichten für Patienten signifikant verbessert.
Literatur:
1 Humbert M et al.: ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022; ehac237 2 Valerio L et al.: Chronic thromboembolic pulmonary hypertension and impairment after pulmonary embolism: the FOCUS study. Eur Heart J 2022; 43(36): 3387-98 3 Lang IM, Madani M: Update on chronic thromboembolic pulmonary hypertension. Circulation 2014; 130(6): 508-18 4 Ghofrani HA et al.: Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013; 369(4): 319-29 5 Sadushi-Kolici R et al.: Subcutaneous treprostinil for the treatment of severe non-operable chronic thromboembolic pulmonary hypertension (CTREPH): a double-blind, phase 3, randomised controlled trial. Lancet Respir Med 2019; 7(3): 239-48
Das könnte Sie auch interessieren:
Funktionsstörung des Myokards: wenn die Entspannung des Herzens gestört ist
Die hypertropheobstruktive Kardiomyopathie (HOCM) ist dadurch charakterisiert, dass die Entspannung des Myokards funktionsgestört ist. Die Folge ist eine zunehmende Verdickung der ...
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...