
Herzinsuffizienz: Daten zur Epidemiologie und Auswirkungen von Kardiomyopathien
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Kardiomyopathien sind nach Bluthochdruck, ischämischen Herzerkrankungen und chronisch-obstruktiven Lungenerkrankungen weltweit die vierthäufigste Ursache für Herzinsuffizienz. Studien, die im Rahmen des European Heart Failure Congress 2023 in Prag präsentiert wurden, beschäftigten sich mit Epidemiologie, Prognose und Auswirkung auf die Lebensqualität Betroffener.
Keypoints
-
Die fünf häufigsten Kardiomyopathien sind dilatative Kardiomyopathien, kardiale Sarkoidose, Myokarditis, hypertrophe Kardiomyopathie (HCM) und Amyloidose.
-
Eine schlechte Prognose haben Amyloidose, arrhythmogene rechtsventrikuläre Kardiomyopathie und linksventrikuläre Non-Compaction-Kardiomyopathie.
-
Bei HCM kommt es derzeit trotz Therapie nur bei 8% der Patienten zur Verbesserung der NYHA-Klasse, bei 23% verschlechtert sich diese sogar.
-
Bei einer HCM ist die Mortalität bei Frauen um den Faktor drei erhöht, bei Männern führte sie zu einer Verdoppelung desRisikos zu sterben.
Prospektive Studie bei Verdacht auf Kardiomyopathien
Kardiomyopathien zählen zu den wichtigsten Ursachen für Herzinsuffizienz und sind eine heterogene Gruppe von Erkrankungen. Es gibt ein weites Spektrum an Kardiomyopathien, jedoch keine prospektive Datenbank, die die Verteilung der Erkrankungen dieser Gruppe umfasst. Diese Lücke wurde nun durch eine Studie am Sahlgrenska CardioMyopathy Prospective Centre (SCMPC) geschlossen, welche die Möglichkeit eröffnet, sämtliche Kardiomyopathien hinsichtlich Phänotyp, Symptomen und Prognose zu vergleichen.
Die SCMPC-Studie1 ist eine prospektive Studie, die Anfang 2018 gestartet wurde und alle Patienten einschließt, die seit diesem Zeitpunkt mit Verdacht auf Kardiomyopathie an das Zentrum zugewiesen wurden. Erfasst wurden Patientencharakteristika, Hintergrund, Familienanamnese, Symptome, diagnostische Untersuchungen und Therapie, konkret: Medikation, Devices und Transplantationen. Blutproben und Biopsien werden in einer Biobank gesammelt. Die Diagnosen erfolgen nach den Kriterien der ESC Working Group on Myocardial and Pericardial Diseases. Endpunkte sind Tod, Herztransplantation oder mechanische Kreislaufunterstützung.
Insgesamt wurden 461 Personen mit einem mittleren Alter von 53,6 Jahren in die Studie eingeschlossen, 73% der Teilnehmer waren Männer. Vorweg – der Verdacht, dass tatsächlich eine Kardiomyopathie vorliegt, wurde in den meisten Fällen erhärtet; lediglich in 9 % der Fälle traf dies nicht zu.
Folgende Kardiomypathien wurden diagnostiziert: 35% waren dilatative Kardiomyopathien (DCM), gefolgt von kardialer Sarkoidose mit 14% und Myokarditis mit 12%. Danach folgten hypertrophe Kardiomyopathien mit 11%, Amyloidose mit 6%, arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) mit 2%, linksventrikuläre Non-Compaction-Kardiomyopathie (LVNC) mit 2% sowie Riesenzell-Myokarditis mit 1%. Zu weiteren 7% summierten sich sehr seltente Kardiomyopathien wie etwa Takotsubo oder peripartale Kardiomyopathie und andere auf.
Patienten mit Myokarditis waren im Schnitt jünger, während Patienten mit kardialer Amyloidose mit einem Altersdurchschnitt von 71 Jahren deutlich älter waren und mehr Komorbiditäten aufwiesen. Bei DCM und Amyloidose war Dyspnoe das häufigste initiale Symptom, ARVC äußerten sich in ventrikulären Arrhythmien. Nach 2,5 Jahren medianem Follow-up waren 86% der Patienten am Leben und nicht transplantiert. Unter den untersuchten Erkrankungen hatte die kardiale Amyloidose ab Einsetzen der Symptomatik die schlechteste Prognose. Als prognostisch ebenfalls ungünstig erwiesen sich ARVC und LVNC.1
Lebensqualität hypertropher Kardiomyopathie
Daten zu Symptomlast und Lebensqualität bei hypertropher Kardiomyopathie liefert eine ebenfalls präsentierte landesweite französische Studie2, in die mehr als 20000 Patienten aufgenommen werden konnten. Kriterium zum Einschluss in die Studie war zumindest eine Hospitalisierung wegen HCM. Bei Einschluss in die Studie hatten 4433 (19%) der Patienten eine obstruktive HCM, im Verlauf der Studie stieg diese Zahl auf 6823 (29%). Das Follow-up betrug im Median 4,4 Jahre, Patienten mit weniger als einem Jahr Follow-up wurden aus der Auswertung ausgeschlossen.
Die Mehrzahl der Studienteilnehmer zeigte sich symptomatisch und war zu 34% in den NYHA-Klassen II bzw. zu 55% in der NYHA-Klasse III. Patienten in höheren NYHA-Klassen wiesen gemessen anhand des Charlson Comorbidity Index Score mehr Komorbiditäten auf. Die Mehrzahl der Studienpatienten (69%) beendete die Studie in derselben NYHA-Klasse, mit der sie in die Studie eingeschlossen wurden. Bei 23% der Patienten kam es zu einer Verschlechterung der NYHA-Klasse, bei acht Prozent kam es zu einer Verbesserung der NYHA-Klasse im Follow-up. Die eingesetzten Therapien zeigt Abbildung 1.2
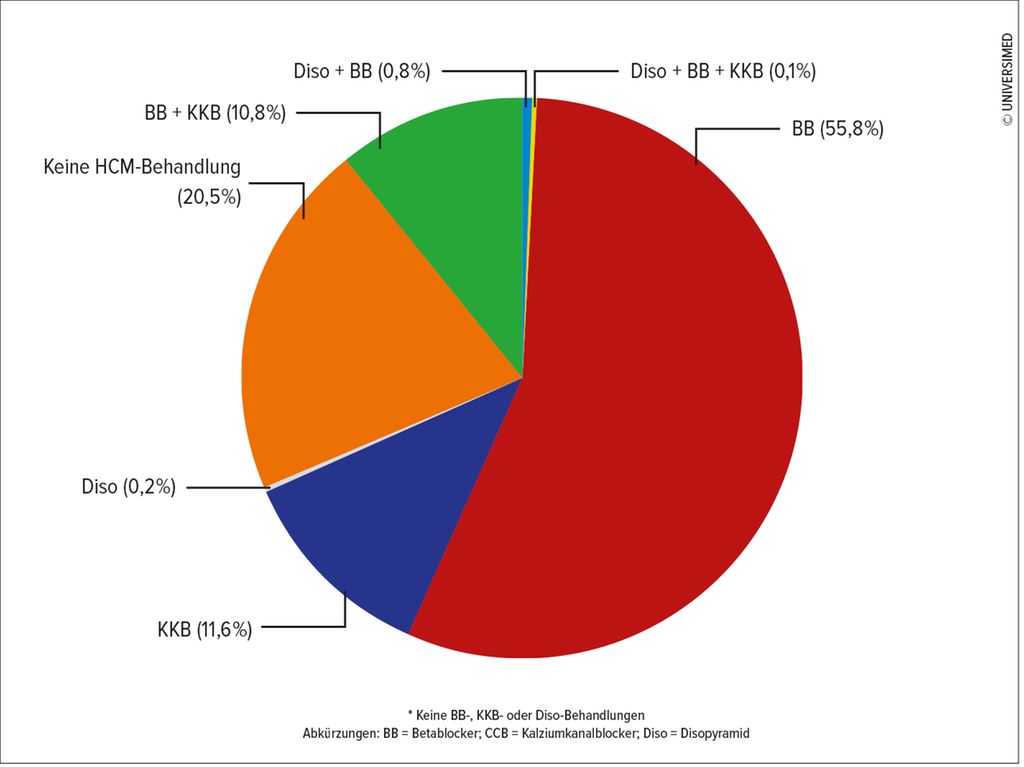
Abb. 1: HCM-Behandlungen (modifiziert nach Charron P et al.)2
Mortalität bei hypertropher Kardiomyopathie
Eine weitere Studie3 untersuchte die Mortalität bei Personen mit HCM und fand im Vergleich zur gesunden Normalbevölkerung eine signifikant erhöhte Gesamtmortalität, wobei diese sich nicht alleine auf das bei HCM erhöhte Risiko für plötzlichen Herztod zurückführen lässt. Es handelt sich bei der Studie um eine retrospektive Kohortenstudie auf Basis mehrerer Datenbanken in Großbritannien und den USA, die Daten von fast 200000 Personen umfasste. Die Patientenpopulation wurde zusätzlich nach Typ der HCM (obstruktiv und nicht obstruktiv) aufgegliedert. Die Auswertung zeigte eine über fast alle Altersgruppen hinweg erhöhte Sterblichkeit. Lediglich bei über 80-Jährigen verschwand diese Assoziation. Aufgrund der niedrigen Mortalität junger Erwachsener trat die Übersterblichkeit bei Personen mit HCM in dieser Altersgruppe besonders hervor. Obstruktive und nicht obstruktive HCM waren mit einem vergleichbaren Mortalitätsrisiko verbunden. Besonders deutlich war der ungünstige Effekt bei Frauen, deren standardisierte Mortalitätsrate um den Faktor drei erhöht war, während HCM bei Männern zu einer Verdoppelung des Risikos zu sterben führte.3
Quelle:
Heart Failure 2023, Sessions: Myocardial Disease - Hypertrophic Cardiomyopathy 1 und Hypertrophic Cardiomyopathy 2, ePosters. 20. bis 23. Mai 2023, Prag
Literatur:
1 Ljungman C et al.: Differences in phenotypes, symptoms and survival in patients with cardiomyopathies, a prospective observational study from the Sahlgrenska Cardiomyopathy Prospective Centre (SCMPC). Presented at Heart Failure 2023 2 Charron P et al.: Clinical burden of obstructive hypertrophic cardiomyopathy: insights from a nationwide study in France. Presented at Heart Failure 2023 3 Tome Esteban M et al.: All-cause mortality in hypertrophic cardiomyopathy (HCM) compared to the general population in England and the US. Presented at Heart Failure 2023
Das könnte Sie auch interessieren:
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...
Immunsuppression und Infektion: Durchimpfen so gut wie möglich
Autoimmunerkrankungen sind mit erhöhtem Infektionsrisiko verbunden, das durch immunsupprimierende Therapien weiter verstärkt wird. Impfungen sind also in der betroffenen Patient: ...