
Einsatz von Vasodilatatoren: PAH assoziiert mit Bindegewebserkrankung
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die pulmonal-arterielle Hypertension (PAH) ist eine seltene Erkrankung, die gemeinsam mit Bindegewebserkrankungen wie der systemischen Sklerose auftreten kann. Die zeitnahe PAH-Diagnose bei diesen Patient:innen ist entscheidend, da sie mit einer erheblichen Prognoseverschlechterung einhergeht. Nach Diagnosestellung mittels invasiver Rechtsherzkatheteruntersuchung werden lungengefäßspezifische, gefäßerweiternde (vasodilatative) Pharmaka eingesetzt.
Keypoints
-
Bei Patient:innen mit Bindegewebserkrankung sollte mind. 1–2-mal pro Jahr mittels nicht-invasiver Untersuchungen (z.B. DETECT-Score) das Risiko für eine PAH bestimmt werden.
-
Bei Verdacht auf aPAH-CTD muss eine frühzeitige Zuweisung an ein Referenzzentrum zur Evaluierung einer PAH mittels Rechtsherzkatheter erfolgen.
-
Bei Diagnose einer aPAH-CTD ist unabhängig vom individuellen Risiko der Beginn einer dualen, oralen Therapie empfohlen.
-
Bei hohem Risiko sollte eine Dreifachtherapie inklusive eines parenteralen Prostacyclins gegeben werden.
Lungenhochdruck (pulmonale Hypertension, PH) ist definiert als eine Erhöhung des invasiv gemessenen mittleren Pulmonalarteriendrucks (mPAP) >20mmHg. Die PH ist eine kardiovaskuläre Erkrankung, die auf die gesamte Weltbevölkerung bezogen sehr häufig ist. Rund 1% der Menschen sind von einer Form der PH betroffen. Es besteht ein eindeutiger Zusammenhang zwischen Höhe des mPAP, Rechtsherzinsuffizienz und -versagen sowie Mortalität.1
Eine pulmonal-arterielle Hypertension (pulmonal-arterielle Hypertonie, PAH) liegt dann vor, wenn neben der Erhöhung des mPAP >20mmHg ein mittlerer Lungenarterienverschlussdruck („mean pulmonary arterial wedge pressure“, mPAWP) ≤15mmHg und ein Lungengefäßwiderstand („pulmonary vascular resistance“, PVR) >2 Wood-Einheiten vorliegen.
Abseits der hämodynamischen Klassifikation gilt Lungenhochdruck als klinisch sehr heterogenes Krankheitsbild. PH ist häufig ein sekundäres Phänomen bei Herz- oder Lungenerkrankung. Jedoch kann eine assoziierte pulmonal-arterielle Hypertension (aPAH: nomenklatorisch eine Unterform der PH) auch im Rahmen einer Bindegewebserkrankung („connective tissue disease“; CTD) entstehen.2 Als Auslöser für eine solche aPAH-CTD gelten Erkrankungen wie die systemische Sklerose (SSc) oder der systemische Lupus erythematodes (SLE).3 Andere Entitäten sind die rheumatoide Arthritis, das primäre Sjögren-Syndrom, die idiopathische entzündliche Myopathie sowie Mischkollagenosen. So tritt beispielsweise bei der SSc eine aPAH bei rund 5–15% der Patient:innen auf. Risikofaktoren sind ein hohes Alter bei Diagnosestellung, limitierte kutane Manifestationen oder Anti-Centromer-Antikörper (ACA). Dabei ist die aPAH-CTD eine seltene Erkrankung („orphan disease“; 5–10/10000). Außerdem ist die Prognose dieser Patient:innen ungleich schlechter als beispielsweise die von Patient:innen mit idiopathischer PAH.4
Diagnostik und Risikoevaluierung
Vor einer invasiven Abklärung soll die Vortestwahrscheinlichkeit einer aPAH bestimmt werden. Für die SSc wurde mit dem DETECT-Score hierbei ein elegantes und im klinischen Alltag leicht anzuwendendes Tool entwickelt.5 In einem ersten Schritt wird auf Basis der Lungenfunktion (forcierte Vitalkapazität und Diffusionskapazität für Kohlenmonoxid), laborchemischer Parameter (proBNP, Harnsäure, ACA), des Vorliegens von Teleangiektasien sowie auf Basis einer Rechtsachsenabweichung im EKG entschieden, ob eine transthorakale Echokardiografie durchgeführt werden muss. Die hierbei gewonnenen Messungen (Flussgeschwindigkeit der Trikuspidalklappeninsuffizienz, rechtsatriale Fläche) bestimmen im zweiten Schritt, ob ein Rechtsherzkatheter indiziert ist.
Die eindeutige Diagnose einer aPAH-CTD beruht auf der Messung mit Rechtsherzkatheter. Dabei ist die aPAH eine prä-kapilläre PH (mPAP >20mmHg und mPAWP ≤15mmHg).1
Spezifische Therapie der aPAH-CTD
Die spezifische Therapie für aPAH-CTD setzt sich aus Wirkstoffen der folgenden drei Substanzklassen zusammen:
-
Endothelin-Rezeptor-Antagonist (ERA)
-
Phosphodiesterase-5-Hemmer (PDE-5-I) bzw. löslicher Guanylatzyklase-Stimulator (sGC)
-
Prostacyclin-Analoga oder Prostacyclin-Rezeptor-Agonist (PRA)
Details zum Handelsnamen, klinisch eingesetzter Dosierung sowie relevanten Zulassungsstudien finden sich in Tabelle 1. Allen angegebenen Substanzen ist gemeinsam, dass sie zu einer signifikanten Reduktion des PVR führen. Dies zeigt sich klinisch in erster Linie in einer Verlängerung der Gehstrecke im 6-Minuten-Gehtest (6MWD) sowie in der klinischen Symptomatik (v.a. Dyspnoe). Einige Studien konnten auch eine signifikante Reduktion relevanter Outcomes (Hospitalisierungen, klinische Verschlechterung, Mortalität) zeigen. Zulassungsstudien erforschen die aPAH-CTD gemeinsam mit anderen Formen der PAH – also auch PAH-assoziiert mit kongenitaler Herzerkrankung oder der idiopathischen PAH. Die aPAH-CTD machte im Schnitt 25–30% aller Patient:innen in der Therapiegruppe aus.
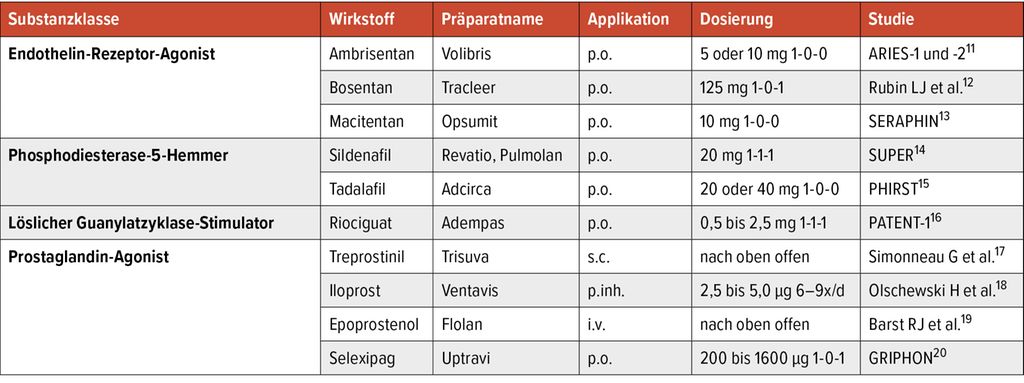
Tab. 1: Vasodilatative Substanzen zur Therapie der aPAH-CTD
Eine initiale Risikostratifìzierung hilft festzulegen, welche initiale Therapie zum Einsatz kommt.1 Einfließende Parameter sind der 6MWD, das Ausmaß der Dyspnoe, die Geschwindigkeit der Krankheitsprogression, erfolgte Synkopen, proBNP, Ergebnisse aus Bildgebungen (Echokardiografie, Magnetresonanztomografie) bzw. die Hämodynamik (Rechtsherzkatheter). Man hat drei Risiko-Strata nach der jährlichen Mortalität festgelegt: niedriges Risiko (<5%), intermediäres Risiko (5–20%) und hohes Risiko (>20%). Nutzte man früher nur einzelne Substanzen, so zeigt sich zunehmende Evidenz für den Vorteil einer initialen Kombinationstherapie. Die Leitlinien fordern bei niedrigem oder intermediärem Risiko den Beginn mit einer dualen oralen Therapie (ERA und PDE-5-I),6 die im Verlauf der Therapie auf eine Dreifachkombination ausgebaut werden kann. Sollte zu Diagnosestellung bereits ein hohes Risiko bestehen, wird zu Therapiebeginn bereits eine Dreifachkombination (ERA, PDE-5-I sowie PRA) gefordert.7
Im Fall relevanter kardiovaskulärer Komorbiditäten neben der aPAH-CTD (dieseKonstellation tritt klinisch immer häufiger auf) ist die derzeitige Datenlage noch unvollständig. Diese Patient:innen waren in den Zulassungsstudien unterrepräsentiert bzw. wurden a priori ausgeschlossen. Zudem sind bei schweren Komorbiditäten (zum Beispiel Niereninsuffizienz, Linksherzinsuffizienz im Rahmen einer Koronar- oder Herzklappenerkrankung oder schwerer Lungenerkrankung mit Fibrose) Wirksamkeit und Verträglichkeit deutlich verringert.8 Daher wird hier nur eine orale Monotherapie mittels ERA oder PDE-5-I empfohlen.1
Klinisch zeitigen diese Therapien häufig nach wenigen Wochen bereits signifikanteEffekte. Nachuntersuchungen sollten zu Beginn mindestens alle drei Monate durchgeführt werden. Auch hier entscheidet das Risiko der Patient:innen über eine eventuelle Therapieeskalation. Dabei braucht es nur wenige Parameter: den 6MWD, proBNP sowie das Ausmaß der Dyspnoe gemessen an der WHOFunctional Class (WHO-FC, synonym mit NYHA-Klassifikation). Anhand eines einfach zu bestimmenden Punkteschemas kann die Therapie eskaliert werden. Sollte unter einer dreifachen, spezifischen Therapie das Risiko jedoch nicht nachhaltig gesenkt und der klinische Verlauf signifikant verbessert werden, sollte rechtzeitig die Evaluierung einer Lungentransplantation erfolgen, obgleich dieses Verfahren aufgrund der systemischen Grunderkrankung außerordentlich komplex ist.
Ausblick und Zusammenfassung
Die Sequenz der aPAH istsekundär bei Bindegewebserkrankungen ein lebensbedrohliches Zustandsbild, was die rechtzeitige Diagnose und aggressive Therapie unabdingbar macht.Aufgrund der niedrigen Prävalenz des Krankheitsbildes wird eine Behandlung in einem Referenzzentrum empfohlen.
Neben den drei bereits etablierten Therapiesäulen befindet sich eine vierte, nicht auf Gefäßerweiterung beruhende Behandlungsoption unmittelbar vor der Marktzulassung: Der Activin-Pfad-Inhibitor Sotatercept wurde in der PULSAR- und STELLAR-Studie erfolgreich bei PAH-Patient:innen eingesetzt.9,10 In beiden Studien wurden rund 60% der Teilnehmenden bereits mit einer Dreifachtherapie behandelt. Eine Zulassung von Sotatercept durch die EMA wird in naher Zukunft erwartet.
Literatur:
1 Humbert M et al.: 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022; 43(38): 3618-731 2 Lau E et al.: Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol 2017; 14(10): 603-14 3 Sanges S et al.: Pulmonary hypertension in connective tissue diseases: What every CTD specialist should know - but is afraid to ask! Rev Med Interne 2024; 45(1): 26-40 4 Le Pavec J et al.: Systemic sclerosis-associated pulmonary arterial hypertension. Am J Respir Crit Care Med 2010; 181(12): 1285-93 5 Coghlan JG et al.: Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7): 1340-9 6 Galiè N et al.: Initial use of Ambrisentan plus Tadalafil in pulmonary arterial hypertension. N Engl J Med 2015; 373(9): 834-44 7 Sitbon O et al.: Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J 2014; 43(6): 1691-7 8 Opitz CF et al.: Pre-capillary, combined, and post-capillary pulmonary hypertension: Apathophysiological continuum. J Am Coll Cardiol 2016; 68(4): 368-78 9 Humbert M et al.: Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 2021; 384(13): 1204-15 10 Hoeper MM et al.: Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med 2023; 388(16): 1478-90 11 Galiè N et al.: Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008; 117(23): 3010-9 12 Rubin LJ et al.: Bosentan therapy for pulmonary arterial hypertension. NEngl J Med 2002; 346(12): 896-903 13 Pulido T et al.: Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013; 369(9): 809-18 14 Galiè N et al.: Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005; 353(20): 2148-57 15 Galiè N et al.: Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009: 119(22): 2894-903 16 Ghofrani HA et al.: Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013; 369(4): 330-40 17 Simonneau G et al.: Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002; 165(6): 800-4 18 Olschewski H et al.: Inhaled iloprost for severe pulmonary hypertension. NEngl J Med 2002; 347(5): 322-9 19 Barst RJ et al.: Acomparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996; 334(5): 296-301 20 Sitbon O et al.: Selexipag for the treatment of pulmonary srterial hypertension. N Engl J Med 2015; 373(26): 2522-33
Das könnte Sie auch interessieren:
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...

(Neue) Blutdruckziele im klinischen Alltag?
Die Diskussion um optimale Blutdruckwerte erhält durch eine aktuelle Analyse aus den USA eine neue Dynamik: Im klinischen Alltag war ein systolischer Blutdruck zwischen 130 und 139mmHg ...