
The molecular biology of sunburn
Sunburn is a common sterile inflammation of the skin caused by excessive exposure to the sun. Surprisingly, the molecular and cellular events underlying the induction of sunburn as well as the long-term consequences are incompletely understood. What happens when sunbeams hit our skin? Why can this result in a painful inflammation? Which cell types and molecular pathways are causing it? The following article tries to give some answers based on publications about irradiated human skin and skin cells.
Keypoints
-
The solar UV spectrum consists of UVA (320-400nm), UVB (280-320nm) and UVC (<280nm).
-
Absorption of UVB by DNA contributes to sunburn in humans.
-
The NLRP1 inflammasome and IL-1 are strong candidates for inducers of sunburn in humans.
-
NLRP1 activation contributes to the development of skin cancer.
Already the ancient Romans characterized inflammation by four cardinal signs, namely redness (rubor), swelling (tumor), warming (calor) and pain (dolor) (Celsus, 25 BC to 50 AD). These are also hallmarks of sunburn, defined as UV (ultraviolet)-induced erythema which is mainly caused by the UVB fraction of solar radiation.1
However, in the recent past we learned that also high doses of UVA emitted by tanning beds induce reddening of the skin.2 Nevertheless, although the dose of UVA originating from the sun and reaching the surface is at least 20-fold higher than that of UVB, the latter is regarded as the most important solar component concerning skin and sunburn.1 UVB is almost completely absorbed by the human epidermis, whereas UVA can also penetrate deeper into the underlying dermis (Fig. 1).

Fig. 1: Absorption of UV radiation by human skin. While the low energetic light wavelengths, comprising infrared (IR) and visible light, are able to pass the ozone layer, roughly 80% of the high-energy components of UV light get absorbed (especially UVC and UVB). Most of the UV radiation hitting our skin is comprised of UVA, which is able to penetrate the epidermis and reach the dermis. Only a portion of UVB radiation hits the skin surface and is absorbed by melanin or urocanic acid. Still, a small fraction that penetrates into the deep layers of the epidermis can cause DNA damage in proliferating keratinocytes of the stratum basale
In contrast, UVC is not relevant for the skin, because it is fully absorbed by the ozone layer, at least at places where this shield is intact.
The good and the bad
Without the sun, no life would exist on earth. Without UVB radiation, epidermal keratinocytes would not be able to convert 7-dehydrocholesterol into the vitamin D precursor previtamin D3, which is essential for calcium metabolism.3 Moreover, through the generation of reactive nitrogen species (RNS), UVA reduces blood pressure, beneficial for an increasing part of the population suffering from hypertension.1 UV-induced pigmentation of the skin (tanning) provides protection from UVB and at least in the western world is still associated with attractiveness.
But even if we acknowledge that the sun has a major impact on our general well-being, its negative effects for the skin clearly predominate. This not only includes sunburn, but also skin aging, mainly caused by UVA-induced generation of reactive oxygen species (ROS) and RNS, and, most importantly, skin cancer.2 Keratinocyte-derived cancer is the most frequent type of malignancy and its incidence is further increasing, mainly due to a longer life expectancy and, partially as a consequence of that, to more exposure of our skin to the sun, particularly to UVB.
Phototherapy for patients suffering from (auto)immune and (auto)inflammatory diseases of the skin is based on UVB-induced immunosuppression and has beneficial effects. However, there is increasing evidence that immunosuppression induced either by sun-derived UVB or by other conditions, such as immunosuppressive drugs for treatment of patients after organ transplantation, is a driving factor of skin cancer development.4
Some general considerations
Mice represent an important model in basic and medical research, including photo-dermatology. However, murine skin is distinct from human skin in several aspects.5 Human epidermis consists of significantly more layers of keratinocytes and total skin is much thicker compared to mice. At the cellular level, murine skin harbors γδ T cells which humans are lacking. Most importantly, mice skin contains numerous hair follicles and the dense fur provides protection from UV radiation. Moreover, mice are nocturnal animals. Therefore, UV radiation does not represent a relevant stressor for mice, although UVB-induced inflammation can be experimentally induced in their skin after shaving off the fur. The key question here is whether UVB-induced inflammation in mice and humans is induced by the same molecular mechanisms.
Human skin lost hair during evolution making it sensitive to radiation by the sun. To compensate for hair loss, human skin might have developed novel protective pathways. Inflammation is essential in all mammals for repair after tissue damage, e.g. after injury or infection.6 Nonetheless, inflammation can also occur without any obvious benefit and underlies the pathology of numerous diseases. Similarly, it is not known whether sunburn in humans is protective, perhaps through experiencing pain and supporting tissue repair. However, the opposite could also be true and sunburn could be part of the problem by inducing or contributing to tissue destruction.
When UVB hits the skin
In principle, the stratum corneum, the outermost layer of the epidermis, protects human skin efficiently from UVB radiation (Fig. 1). Melanin, produced by melanocytes upon UVB irradiation and handed over to keratinocytes, absorbs UVB and accumulates in the corneum.7 A second strong absorber of UVB is urocanic acid, a degradation product of the amino acid histidine and of the histidine-rich protein filaggrin, expressed in the granular layer directly below the stratum corneum.4
Furthermore, DNA is also an excellent chromophore for UVB. Almost 40 years ago, it was demonstrated that the UV action spectrum for erythema formation in human skin is identical to the absorption spectrum of DNA corrected for the optical effects of the stratum corneum.8 While not being a conclusive evidence, this correlation strongly suggests that UVB induces sunburn via its absorption by DNA.
In fact, genomic and mitochondrial DNA of epidermal keratinocytes absorbs the fraction of UVB passed through the corneum causing energetic excitation of the DNA (Fig. 2). In most cases, this energy is simply released as heat, but rarely the excited state of DNA can also induce formation of photolesions, such as cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6–4) pyrimidones.1 In addition, UVA-induced ROS cause single strand breaks (SSB) and 8-oxo-7,8-dihydroguanine formation.3
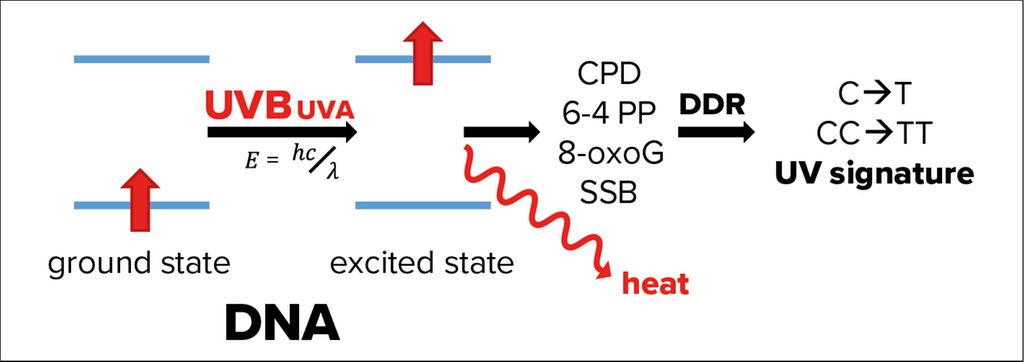
Fig. 2: Effects of UVA and UVB radiation on DNA. If not released as heat, energetic excitation of DNA through the absorption of UVB radiation can cause photolesions of the DNA, like the formation of cyclobutane pyrimidine dimers (CPDs), pyrimidine (6-4) pyrimidones (6-4 PP). Absorption of UVA causes mainly 8-oxo-7,8-dihydroguanines (8-oxoG), as well as single strand breaks of the DNA (SSB). These photolesions activate DNA damage response (DDR) pathways, which, in rare cases, make mistakes leaving a so-called UV signature marked by single- or double cytosine-to-thymidine mutations (C->T or CC->TT) at di-pyrimidine sites
Photolesions activate an efficient network of pathways, which sense these DNA modifications, signal to transducers, stop cell division and repair the damage through highly conserved genome maintenance pathways, collectively termed DNA damage response (DDR).9 The fact that an exposure of 1 hour to sunlight causes 100000 DNA lesions per cell demonstrates the essential role of the DDR in epithelial keratinocytes.10 Research of the last years shows that the DDR is not restricted to the nucleus but connected to several other cell-autonomous and systemic pathways.10
Although the DDR is very efficient, not all DNA modifications can be correctly repaired. Therefore, UVB leaves its marks in the DNA as the so-called UV signature, characterized by C (cytosine) to T (thymidine) and CC to TT mutations at di-pyrimidine sites caused by incorrectly repaired CPDs.11 90% of keratinocyte-derived cancers (basal and squamous cell carcinoma, BCC and SCC) are caused by UV radiation.1 Although compared to melanoma these carcinomas rarely metastasize and are fatal, they are associated with high morbidity and a significant burden for the public health system.
TNF-α or IL-1?
Tumor necrosis factor α (TNF-α) and interleukin-1α/β (IL-1) represent powerful pro-inflammatory cytokines, which can initiate an inflammatory response.12,13 Experiments demonstrated that UVB-induced inflammation of mouse ears is dependent of toll-like receptor 3 (TLR3) expression (Fig. 3).14 This pattern recognition receptor is expressed by keratinocytes and activated by double stranded (ds)RNA, which is associated with viral infection, but does usually not occur endogenously in mammalian cells. It was revealed that UVB modifies the short U1 spliceosomal RNA in a way that it can activate TLR3 and in turn induce TNF-α expression and secretion, thereby initiating an inflammatory response. These fascinating results support the observation that sunburn is linked to UVB absorption by (deoxy)ribonucleic acid.8 As TLR3 is located on the cell surface, its activation by UVB-modified U1 RNA requires lysis of keratinocytes. However, UVB-induced lysis of epidermal keratinocytes takes significantly longer than UVB-induced erythema formation. Therefore, it is not clear whether the described pathway is indeed physiologically relevant for sunburn in humans.
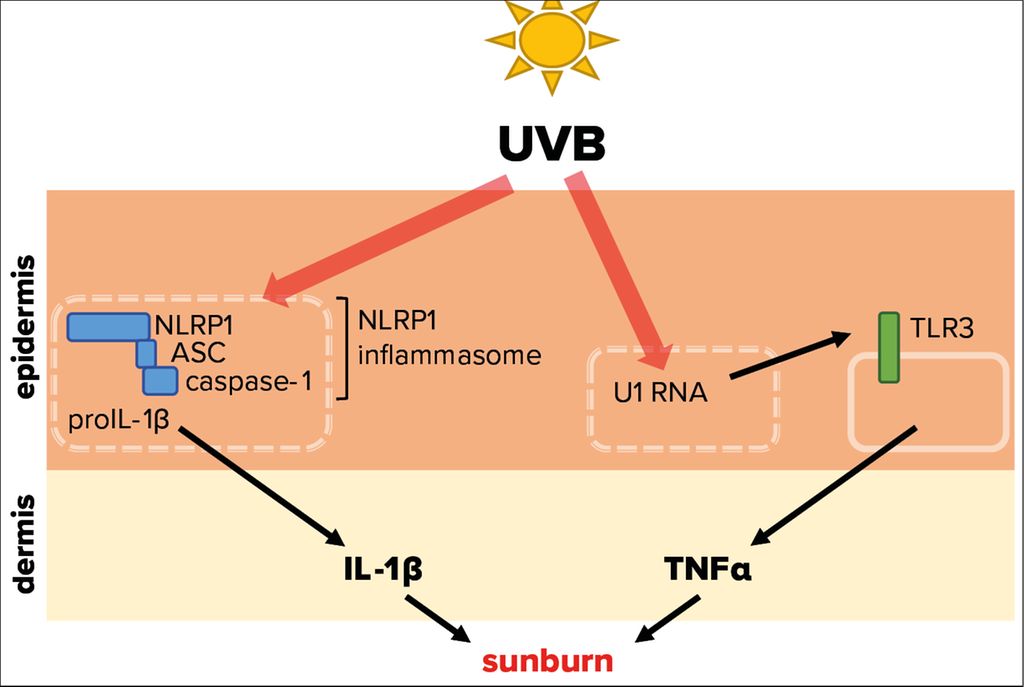
Fig. 3: Molecular mechanisms triggered by UVB radiation leading to sunburn. UVB radiation that penetrates the skin is able to activate NLRP1, a cytosolic pattern recognition receptor of the innate immune system. This leads to the assembly of the NLRP1 inflammasome, comprising of adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1, resulting in activation and secretion of the proinflammatory cytokine IL-1β. In addition, UVB-modified U1 spliceosomal RNA from dead or dying cells can trigger the activation of the membrane-bound toll-like receptor 3 (TLR3), causing secretion of the proinflammatory cytokine TNFα. Both cytokines are therefore potentially involved in the sterile UVB-induced inflammation of the skin, termed sunburn
Irradiation of human primary keratinocytes with a physiological dose of UVB causes secretion of IL-1α and -β15 suggesting that IL-1 induces or contributes to sunburn in humans.16 ProIL-1β requires activation by the protease caspase-1, which in turn is activated upon assembly of innate immune complexes, termed inflammasomes (Fig. 3).17
In human keratinocytes, NLRP1 is the principal inflammasome sensor18 and ablation of NLRP1 expression in these cells prevents proIL-1β activation by UVB.19 Interestingly, patients with gain-of-function mutations of NLRP1 suffer from inflammatory skin syndromes suggesting that NLRP1 activation in epidermal keratinocytes causes skin inflammation. Furthermore, these patients are predisposed for the development of SCC.18 As repeated or chronic NLRP1 inflammasome activation in epidermal keratinocytes upon expression of hyperactive mutants favors the development of SCC, it is also likely that sunburn, if indeed induced by NLRP1 activation and downstream IL-1 secretion, supports skin cancer development. However, it is currently unknown how UVB activates NLRP1 at the molecular level. Very recently, it was demonstrated that long dsRNA activates NLRP1 in human keratinocytes.20 As the dsRNA receptor TLR3 is activated upon binding of U1 RNA after its modification by UVB,14 a similar mechanism might underlie UVB-induced NLRP1 activation.
It should be noted that the NLRP1 pathway is not conserved between human and mice. Although UVB-induced inflammation in the shaved back skin of mice is caspase-1- and IL-1β-dependent, an unknown immune cell type carries out this task in murine skin.15,21 In contrast to human primary keratinocytes, murine keratinocytes do neither express significant amounts of NLRP1 nor of proIL-1β.21
Literature:
1 Young AR et al.: J Am Acad Dermatol 2017; 76(3S1): S100-9 2 Krutmann J, Berneburg M: Hautarzt 2021; 72(1): 2-5 3 Schuch AP et al.: Free Radic Biol Med 2017; 107: 110-24 4 Bernard JJ et al.: Nat Rev Immunol 2019; 19(11): 688-701 5 Nestle FO et al.: Nat Rev Immunol 2009; 9(10): 679-91 6 Medzhitov R: Nature 2008; 454(7203): 428-35 7 D‘Orazio J et al.: Int J Mol Sci 2013; 14(6): 12222-48 8 Parrish JA et al.: Photochem Photobiol 1982; 36(2): 187-91 9 Jackson SP, Bartek J: Nature 2009; 461(7267): 1071-8 10 Chatzidoukaki O et al.: . Trends Genet 2020; 36(10): 777-91 11 Cadet JT et al.: Photochem Photobiol 2015; 91(1): 140-55 12 Bradley JR: J Pathol 2008; 214(2): 149-60 13 Mantovani A et al.: Immunity 2019; 50(4): 778-95 14 Bernard JJ et al.: Nat Med 2012; 18(8): 1286-90 15 Feldmeyer L et al.: Curr Biol 2007; 17(13): 1140-5 16 Faustin B, Reed JC: Trends Cell Biol 2008; 18(1): 4-8 17 Broz P, Dixit VM: Nat Rev Immunol 2016; 16(7): 407-20 18 Zhong FL et al.: Cell 2016; 167(1): 187-202.e17 19 Fenini G et al.: J Invest Dermatol 2018; 138(12): 2644-52 20 Bauernfried S et al.: Science 2021; 371(6528) 21 Sand J et al.: Cell Death Dis 2018; 9(2): 24
Das könnte Sie auch interessieren:
Supportive Onkodermatologie 2026
Der vermehrte Einsatz von Tumor-Immuntherapien hat in den letzten Jahren zu einem grundlegenden Wandel in der Onkologie geführt. Während Immuncheckpoint-Inhibitoren die ...

Klimawandel und neue Infektionskrankheiten im Blick behalten
Aufgrund des Klimawandels wurde in letzter Zeit ein deutlicher Anstieg neu auftretender Infektionskrankheiten in Europa verzeichnet. Dies führt zu neuen Herausforderungen sowohl bei der ...
A proven standard in hyperhidrosis treatment
Primary hyperhidrosis significantly impairs quality of life. This article reviews the role of botulinum toxin A as a safe and effective therapy, highlights proper patient assessment with ...