Corneodesmosomen und ihre Rolle bei Peeling-Skin-Erkrankungen
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Im Stratum corneum bilden die ineinander verhakten und durch Corneodesmosomen fest verbundenen Korneozyten ein stabiles Gerüst für interzelluläre Barrierelipide. Die physiologische Hautabschuppung setzt einen sukzessiven Abbau der verbindenden Proteinstrukturen voraus und wird durch Proteasen und Proteaseinhibitoren reguliert. Bei Strukturdefekten oder Dysbalancen des Regulationssystems kommt es zur unkontrollierten Desquamation, dem sogenannten „Peeling“. Über die verschiedenen Peeling-Skin-Erkrankungen gibt der folgende Artikel einen Überblick.
Keypoints
-
Der sukzessive Abbau der Corneodesmosomen spielt für die physiologische Desquamation eine essenzielle Rolle.
-
Fehlende Proteaseinhibitoren oder hereditäre Strukturdefekte der Corneodesmosomen führen klinisch zur vermehrten Hautabschuppung, dem sogenannten „Peeling“.
-
Peeling-Skin-Erkrankungen werden in die lokalisierte, die generalisierte nicht inflammatorische und die generalisierte inflammatorische Form unterteilt, wobei Letztere den schwersten Verlauf zeigt.
-
Die Therapie orientiert sich am Schweregrad der Hautentzündung, assoziierten Symptomen und dem Leidensdruck der Betroffenen.
Das Stratum corneum der Epidermis stellt die erste gegenüber der Außenwelt separierende Barrierestruktur dar. Es besteht aus mehreren Lagen von ineinander verhakten und durch Corneodesmosomen (CDs) fest verbundenen Korneozyten, die ein stabiles Gerüst für die interzelluläre lipidreiche Extrazellulärmatrix formen. Jeder Korneozyt wird durch ca. 1000 CDs mit anderen Korneozyten verbunden. CDs, die sich aus Desmosomen des Stratum granulosum transformieren, sind Transmembranproteine bestehend aus Desmoglein 1, Desmocollin 1 und Corneodesmosin. Sie werden mit Polypeptiden wie Plakoglobin vernetzt und daneben auch enzymatisch in die „cornified envelopes“ der Korneozyten eingebaut. Dabei spielen die kalziumabhängigen Transglutaminasen 1 und 5 eine wichtige Rolle.
Die physiologische Abschuppung der äußersten Hornschicht setzt einen sukzessiven Abbau der CDs voraus und wird durch ein fein balanciertes System von Serinproteasen (Kallikreine) und Cysteinproteasen (Kathepsine) einerseits und Proteaseinhibitoren (Cystatin A, Calpastatin, SERPINB8 und LEKTI) andererseits reguliert (Abb. 1). Die Aktivität der Proteasen wird dabei durch einen neutral-alkalischen pH-Wert erhöht. Eine Dysbalance dieses komplexen Regulationssystems mit einem Mangel an Proteaseinhibitoren oder Strukturdefekten der CDs führen zu einer vermehrten, unkontrollierten Desquamation, was sich klinisch als feine Schuppung („Peeling“) manifestiert (Abb. 1).
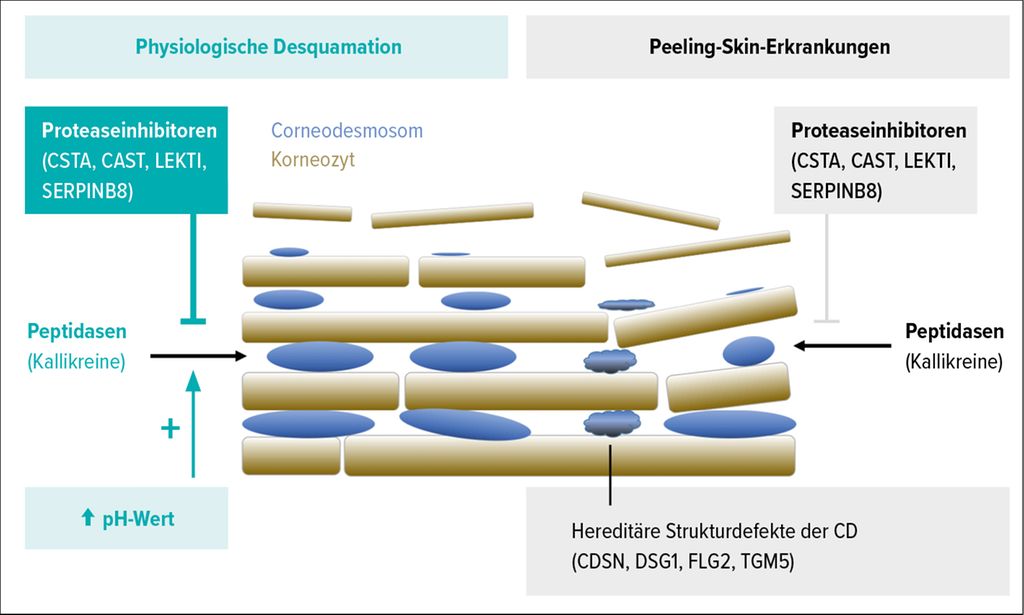
Abb. 1: Physiologische Desquamation versus Peeling-Skin-Erkrankungen
Peeling-Skin-Erkrankungen stellen eine heterogene Gruppe sehr seltener, autosomal-rezessiv vererbter Verhornungsstörungen dar, die klinisch durch eine oberflächliche Schuppung bis Blasenbildung charakterisiert sind. Häufige Trigger, die zur Verschlechterung der Peeling-Skin-Erkrankungen (PSE) führen, sind mechanische und thermische Reize sowie längerer Wasserkontakt. Ursächlich liegen den PSE angeborene Strukturdefekte der CDs, eine gestörte Verankerung der CDs in den „cornified envelopes“ der Korneozyten durch defekte Transglutaminasen oder ein Mangel an Proteaseinhibitoren zugrunde.
Lokalisierung und Bezeichnung
PSE werden in lokalisierte (akrale), generalisierte nicht inflammatorische und generalisierte inflammatorische Subtypen unterteilt (Tab. 1), wobei Letztere den schwersten Verlauf zeigen und neben Erythrodermie auch mit Pruritus, Atopie und Gedeihstörung assoziiert sind.
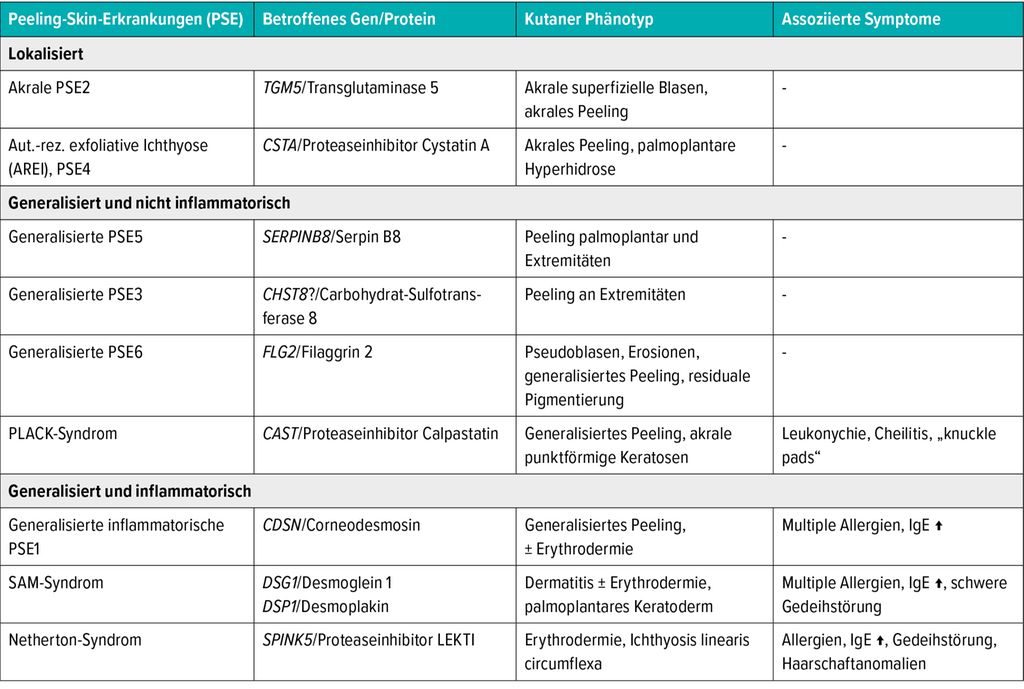
Tab. 1: Einteilung der Peeling-Skin-Erkrankungen
Namentlich erstmals erwähnt wurde „the peeling skin syndrome“ 1982 von Levy und Goldsmith. Die Erstbeschreibung geht jedoch auf das Jahr 1924 zurück, in dem Wile eine Familie mit drei ungewöhnlichen Fällen einer kongenitalen ichthyosiformen Erythrodermie beschrieb. Der ursprüngliche und immer noch häufig verwendete Begriff „Peeling-Skin-Syndrom“ ist irreführend, weil die lokalisierten PSE und die generalisierten nicht inflammatorischen Subtypen mit Ausnahme des PLACK-Syndroms nur eine Hautbeteiligung ohne weitere Symptome zeigen und somit keine Syndrome darstellen. Daher sollte der Name „Peeling-Skin-Erkrankung“ favorisiert werden.
Das klinische Bild
Die generalisierte nicht inflammatorische PSE5 beginnt meist im 3. Lebensjahr mit Peeling an den Unterarmen, Beinen und palmoplantar. Sie wird durch biallelische Mutationen im Gen SERPINB8 verursacht, wodurch es zum Funktionsverlust des Serinproteaseinhibitors (Serpin) und zu einem vermehrten Abbau der CDs kommt.
Bei der PSE3 zeigt sich ab dem Kindesalter ein Peeling an den Extremitäten. Zur PSE3 sind lediglich Fälle aus einer einzigen pakistanischen Familie publiziert. Als ursächlich wurden von den Autoren Mutationen im Carbohydrat-Sulfotransferase- 8-Gen, das eine Rolle bei der Verankerung der CDs spielen könnte, angenommen. Die besagten Varianten in CHST8 wurden allerdings in einer Folgearbeit als Polymorphismus identifiziert, sodass die Krankheitsursache letztlich unklar bleibt.
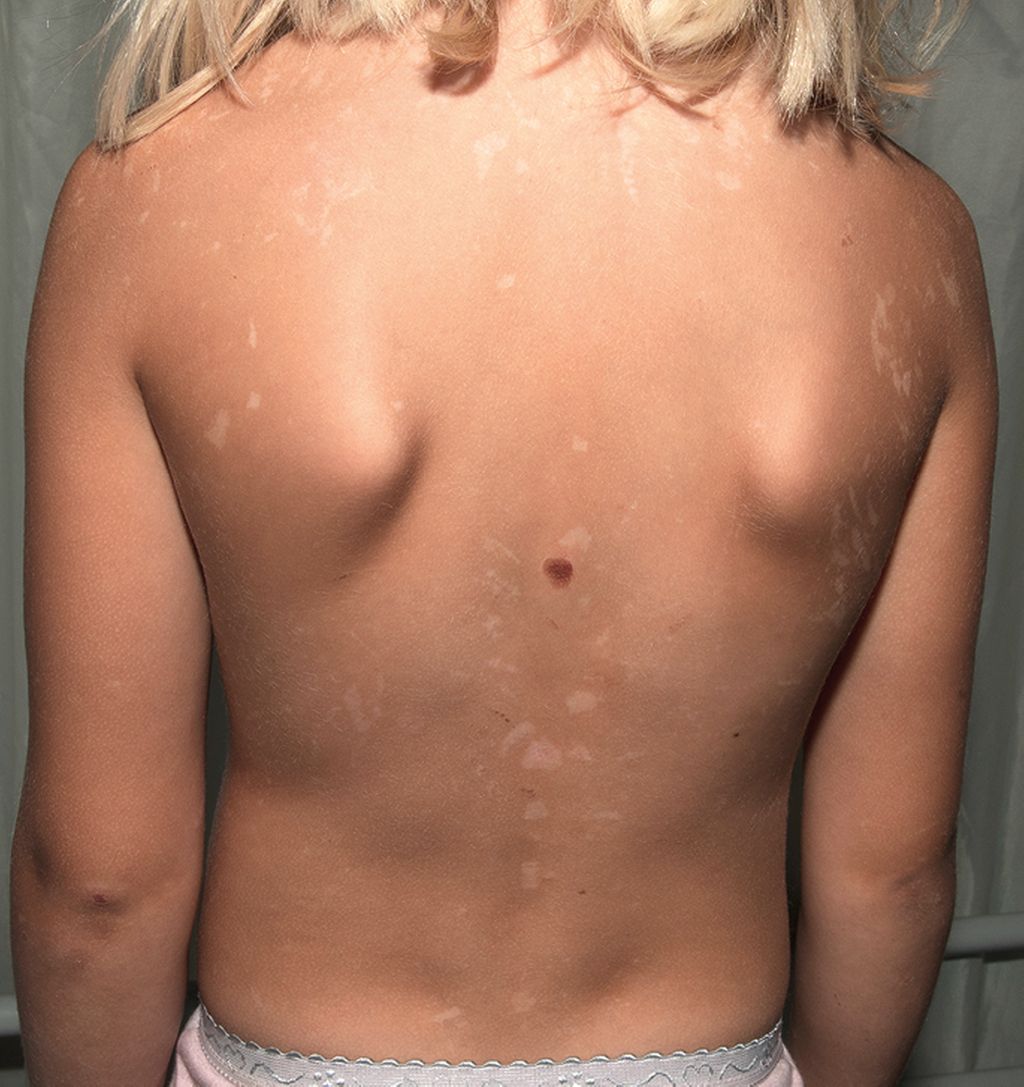
Abb. 2: Residuale Hypopigmentierungen und Blase bei einem Mädchen mit generalisierter PSE6
Die häufigste generalisierte nicht inflammatorische PSE ist die PSE6, bei der die betroffenen Kinder neben Peeling auch pseudobullöse Läsionen und Erosionen nach minimalen Traumata aufweisen. Nach Abheilung werden in der Literatur residuale Hyperpigmentierungen dokumentiert, wobei wir bei einem Mädchen an unserem Expertisezentrum auch residuale Hypopigmentierungen sehen (Abb. 2). Mit zunehmendem Patientenalter verbessert sich der Phänotyp, sodass Erwachsene nur noch milde Hautveränderungen aufweisen. Der PSE6 liegen biallelische Mutationen im FLG2-Gen zugrunde. Es codiert für Filaggrin 2, das im Stratum granulosum gebildet wird und sich im gesamten Stratum corneum findet. Interessanterweise kolokalisiert es mit Corneodesmosin (CDSN). In der Vergangenheit wurde gezeigt, dass das Fehlen von FLG2 mit einer signifikant verminderten CDSN-Expression einhergeht. Somit ist FLG2 essenziell für die Zell-Zell-Adhäsion im Stratum corneum.
Syndromale Formen
PLACK-Syndrom
Das Akronym PLACK setzt sich aus Peeling skin, Leukonychie, Akrale punktförmige Keratosen, Cheilitis und Knuckle pads zusammen. Das Syndrom dieser Symptomatik wird durch Mutationen in CAST verursacht, wodurch der Proteaseinhibitor Calpastatin fehlt und es zu einem frühzeitigen und vermehrten Abbau der CDs kommt. Die PSE1 manifestiert sich bei Geburt und erinnert an eine ichthyosiforme Erythrodermie, die meist auch noch im Erwachsenenalter besteht. Es kommt zu einem lebenslangen generalisierten Peeling, teilweise auch Erosionen, nur die Handflächen und Fußsohlen bleiben ausgespart. Die Betroffenen haben oft einen ausgeprägten Juckreiz, Nahrungsmittelallergien und Asthma bronchiale. Das Gesamt-IgE im Serum ist deutlich erhöht. Histologisch findet sich eine junktionale Spaltbildung zwischen Stratum granulosum und Hornschicht. Die PSE1 wird durch biallelische Mutationen in CDSN verursacht, was zu defekten CDs und einem fehlenden Zusammenhalt der Korneozyten führt.
SAM-Syndrom
Dieses Syndrom – SAM ist ein Akronym aus Schwere Dermatitis, multiple Allergien und Metabolisches Verlust-Syndrom – weist eine phänotypische Heterogenität auf. Als Ursache wurden initial biallelische Mutationen im Desmoglein-Gen (DSG1) identifiziert, die zu defekten und fehlenden CDs sowie Desmosomen führen. In rezenten Publikationen wurden auch Patienten mit SAM-Syndrom und heterozygoten Mutationen im Desmoplakin-Gen (DSP1) beschrieben.
Netherton-Syndrom
Dieses manifestiert sich bei Geburt mit einer ichthyosiformen Erythrodermie, im Kindesalter tritt dann die charakteristische Ichthyosis linearis circumflexa auf. Neben einer Gedeihstörung und rezidivierenden Infekten im Säuglings- und Kleinkindesalter ist das Netherton-Syndrom durch Allergien, ein stark erhöhtes Gesamt-IgE und eine vermehrte Brüchigkeit der Haare durch Haarschaftanomalien (v.a. Trichorrhexis invaginata) gekennzeichnet. Es besteht ein stark erhöhtes Risiko für Basalzell- und Plattenepithelkarzinome. Der Erkrankung liegen biallelische Mutationen in SPINK5 zugrunde, welches für den Proteaseinhibitor LEKTI codiert. Durch die verminderte Inhibierung von Serinproteasen wie Kallikrein 5 findet ein beschleunigter Abbau der CDs statt und es kommt in weiterer Folge zum Peeling der oberen Stratum-corneum-Schichten.
Wichtige Differenzialdiagnosen
Die generalisierten PSE sind von der autosomal-rezessiven kongenitalen Ichthyose, der hypohidrotischen ektodermalen Dysplasie und der atopischen Dermatitis zu unterscheiden. Die akrale PSE2 ist neben Peeling an Fingern, Zehen und Fersen auch durch akrale superfizielle Blasen charakterisiert. Sie wird durch Mutationen im TGM5-Gen verursacht. Als Folge des Mangels an Transglutaminase 5 kommt es zu einer fehlerhaften Fixierung der CDs an den „cornified envelopes“.
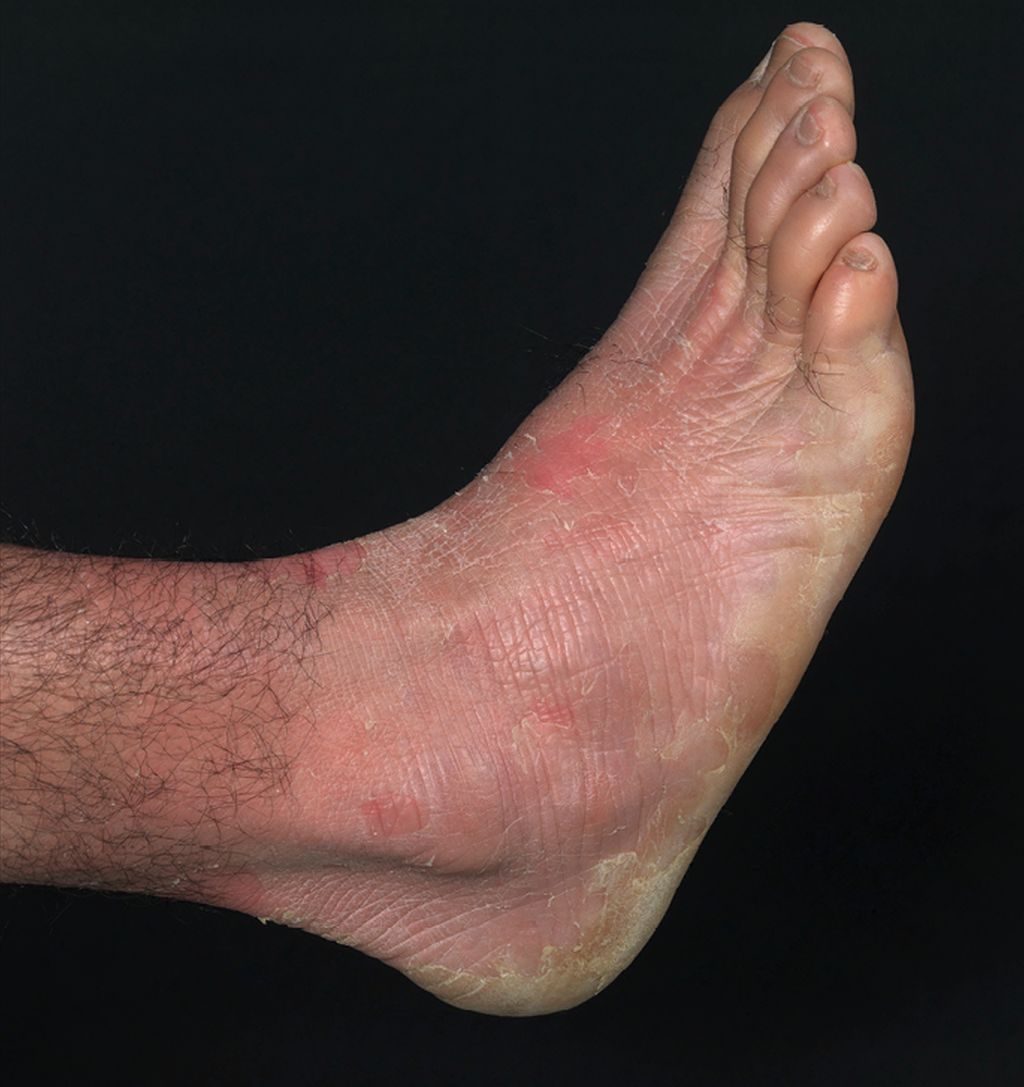
Abb. 3: Akrales Peeling bei einem Jugendlichen mit AREI
Die akrale PSE4 ist besser bekannt unter dem Namen autosomal-rezessive exfoliative Ichthyose (AREI). Klinisch findet sich ein akrales Peeling, häufig besteht eine palmoplantare Hyperhidrose (Abb. 3). Biallelische Mutationen im CSTA-Gen führen zu einem Mangel des Proteaseinhibitors Cystatin A und wiederum in weiterer Folge zum beschleunigten Abbau der CDs.
Wichtige Differenzialdiagnosen der lokalisierten PSE sind die Epidermolysis bullosa simplex und die superfizielle epidermolytische Ichthyose.
Behandlung und Fazit
Die Therapie der PSE orientiert sich am Schweregrad der Hautentzündung, assoziierten Symptomen und dem Leidensdruck der Betroffenen. Eine Basispflege mit Glycerol oder Urea-haltigen Cremen oder Salben wird empfohlen. Trauma und Friktion sowie weitere Triggerfaktoren wie Hitze und längere Nässe sollten vermieden werden. Topische Proteasehemmer (z.B. Atopiclair® Creme) und leicht saure Externa (z.B. Eucerin® pH5 Lotion) können zur Verbesserung des Peelings führen, indem sie die Aktivität der Proteasen im Stratum corneum abschwächen. Bei inflammierter Haut werden topische Kortikosteroide am besten proaktiv eingesetzt. Systemtherapien mit Dupilumab oder Secukinumab werden erfolgreich bei den generalisierten inflammatorischen PSE eingesetzt.
PSE stellen eine heterogene Gruppe von Verhornungsstörungen dar (Tab.1). Klinisch sollte bei oberflächlicher feiner Schuppung („Peeling“) und superfiziellen Blasen an Händen und Füßen differenzialdiagnostisch an eine PSE gedacht und eine entsprechende molekulargenetische Untersuchung erwogen werden.
Literatur:
beim Verfasser
Das könnte Sie auch interessieren:
Supportive Onkodermatologie 2026
Der vermehrte Einsatz von Tumor-Immuntherapien hat in den letzten Jahren zu einem grundlegenden Wandel in der Onkologie geführt. Während Immuncheckpoint-Inhibitoren die ...

Klimawandel und neue Infektionskrankheiten im Blick behalten
Aufgrund des Klimawandels wurde in letzter Zeit ein deutlicher Anstieg neu auftretender Infektionskrankheiten in Europa verzeichnet. Dies führt zu neuen Herausforderungen sowohl bei der ...
A proven standard in hyperhidrosis treatment
Primary hyperhidrosis significantly impairs quality of life. This article reviews the role of botulinum toxin A as a safe and effective therapy, highlights proper patient assessment with ...