
Neue Therapieansätze und Ausbau bestehender Konzepte
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Vom 7. bis 11.10.2025 fand in Wien der 30. Kongress der World Muscle Society statt. Neben den neuesten Studien und Daten stand auch der persönliche Austausch zwischen den rund 15000 Teilnehmer:innen aus 80 verschiedenen Ländern im Vordergrund. Vor allem in den Therapiefeldern der spinalen Muskelatrophie, der Duchenne-Muskeldystrophie und der myotonen Dystrophie gab es vielversprechende Studienergebnisse, die bestehende Behandlungskonzepte verbessern und erweitern könnten.
Lange gab es – abseits von Kortikosteroiden – für Patient:innen mit Muskelkrankheiten nur wenige Therapieoptionen. In den letzten Jahren kam mit zielgerichteten Therapien reichlich Bewegung in das Feld. So wurden heuer beim Kongress der World Muscle Society in Wien wieder einige Highlights vorgestellt, die Hoffnung für Betroffene bieten können.
Update: spinale Muskelatrophie
Überzeugende 3-Jahres-Daten zu Risdiplam bei Säuglingen mit präsymptomatischer SMA
Bei der spinalen Muskelatrophie (SMA) ist ein frühzeitiger Behandlungsbeginn entscheidend für den Therapieerfolg, jedoch beginnt die Degeneration der Motoneurone bereits vor dem Auftreten erster Symptome.1 Ursächlich für die Erkrankung ist eine Mutation des „Survival motor neuron“-1-Gens (SMN1). Einer der Behandlungsstandards ist Risdiplam. Das Medikament modifiziert das Spleißen der sonst unfunktionalen, aber potenziell kompensatorischen SMN2-mRNA und ermöglicht somit die Bildung eines funktionalen SMN-Proteins.2,3
Patient:innen, die im Säuglingsalter an SMA erkranken, sind oft unfähig, frei zu sitzen oder zu rollen. „Die RAINBOWFISH-Studie ist eine einarmige Phase-II-Studie zur Bewertung der Wirksamkeit, Sicherheit, Pharmakokinetik und -dynamik von Risdiplam bei Säuglingen (von der Geburt bis zu 6 Wochen, n=25) mit genetisch diagnostizierter präsymptomatischer SMA“, erläuterte Prof. Dr. Maria Mazurkiewicz-Bełdzinska von der Medizinischen Universität in Gdansk, Polen.4 Der primäre Studienendpunkt wurde bereits nach 12 Monaten erreicht: So konnten 80% der Patient:innen mit zwei SMN2-Genkopien für mehr als 5 Sekunden ohne Unterstützung sitzen.5
Erreichen und Beibehaltung motorischer Meilensteine über 3 Jahre unter Risdiplam
Aktuelle 3-Jahres-Daten zeigten, dass 91% der Teilnehmer:innen die wichtigsten motorischen Meilensteine wie Sitzen ohne Unterstützung, Stehen und Gehen erreichen konnten und diese Fähigkeiten beibehielten. Sie behielten zudem ihre bulbäre Funktion bei und benötigten – sofern kein Infekt vorlag – keine respiratorische Unterstützung.
RAINBOWFISH ist die erste klinische Studie, die auch eine kognitive Beurteilung durchführte. Kinder, die 3 Jahre lang mit Risdiplam behandelt wurden, zeigten ähnliche kognitive Fähigkeiten wie normal entwickelte Kinder ohne SMA. Die Daten waren über alle SMN2-Kopienzahlgruppen hinweg ähnlich. In puncto Sicherheit wurden während der dreijährigen Behandlungszeit keine schweren Nebenwirkungen dokumentiert.4,5
DEVOTE-Studie Teil C: Therapienutzen unter erhöhter Nusinersen-Dosis
„Für den optimalen Therapieerfolg sollte eine SMA-Behandlung mit Nusinersen insbesondere bei Säuglingen und Kleinkindern so früh wie möglich begonnen werden“, leitete Prof. Dr. Eugenio Mercuri von der Katholischen Universität in Rom, Italien, seinen Vortrag ein.6
Die dreiteilige Phase-II/III-Studie DEVOTE untersuchte den Nutzen und das Sicherheitsprofil der höheren Nusinersen-Dosierung 50/28mg.7 An Teil C der Studie nahmen Patient:innen (n=40) mit infantiler oder später auftretender SMA teil, die vor Studienbeginn ≥1 Jahr lang 12/12mg Nusinersen erhalten hatten. Insgesamt waren 40% der Studienteilnehmer:innen <18 Jahre und 60% >18 Jahre alt, sie wurden im Median bereits 3,9 Jahre mit 12/12mg Nusinersen behandelt. Die Teilnehmer:innen erhielten vier Monate ±14 Tage nach ihrer letzten 12-mg-Dosis eine Anfangsdosis von 50mg, gefolgt von zwei Erhaltungsdosen in Höhe von 28mg in einem Abstand von 4 Monaten.8
Motorische Verbesserungen unter Nusinersen 50/28g
Die Teilnehmer:innen zeigten nach Umstellung auf 50/28mg Nusinersen nach 302 Tagen Verbesserungen der motorischen Funktionen (Abb. 1): Der mittlere Anstieg gegenüber dem Ausgangswert betrug in der Hammersmith Functional Motor Scale Expanded (HFMSE) +1,8 Punkte und im Revised Upper Limb Module (RULM) +1,2 Punkte. Die Verbesserungen traten in einem Zeitraum auf, in dem die Therapieerfolge mit der 12/12mg-Standarddosis nach anfänglich deutlichen Verbesserungen üblicherweise in ein Plateau übergehen. 50/28mg Nusinersen wurde im Allgemeinen gut vertragen, wobei die Sicherheitssignale weitgehend dem Profil der 12/12-mg-Dosierung entsprachen.8 Die Studienergebnisse sprechen somit für einen Wechsel von der 12/12-mg- zur 50/28-mg-Dosierung.

Abb. 1: 53% der Teilnehmer:innen der DEVOTE-Studie hatten eine verbesserte motorische Fuktion unter erhöhter Nusinersen-Dosis. Die Verbesserungen wurden anhand der Hammersmith Functional Motor Scale Expanded (HFMSE) nach 302 Tagen unter Nusinersen 50/28mg erfasst7
Update: Duchenne-Muskeldystrophie (DMD)
Bei der DMD liegt eine Mutation des X-chromosomal-rezessiv vererbten Dystrophingens vor. Das Dystrophinprotein spielt eine essenzielle Rolle als Strukturprotein der Skelettmuskeln. Die Erkrankung betrifft daher fast nur Jungen.
Unzureichende Dystrophinspiegel unter dem Exon-51-Skipping-Faktor PGN-EDO51
Prof. Dr. Hugh McMillan vom Children’s Hospital of Eastern Ontario in Ottawa, Kanada, stellte die Daten der Phase-II-Studie CONNECT1-EDO51 vor. Diese untersuchte die Wirksamkeit von PGN-EDO51, einem Wirkstoff, der ein Exon-51-Skipping und somit die Herstellung eines verkürzten, funktionalen Dystrophins ermöglicht. Die „Enhanced delivery oligonucleotide“-Technologie (EDO) optimiert hierbei die Gewebezufuhr und die nukleäre Aufnahme des Wirkstoffes.9 Nichtklinische Studien hatten im Vorfeld gezeigt, dass eine einzelne PGN-EDO51-Dosis bereits hohe Exon-Skipping-Raten und einen Anstieg der Dystrophinlevel erzielen konnte.10
In der CONNECT1-EDO51-Studie erhielten DMD-Patient:innen vier PGN-EDO51-Dosen über 3 Monate in einer Konzentration von 5mg/kg (n=3) oder 10mg/kg (n=4). Das ermittelte Sicherheitsprofil war günstig: Unter 5mg/kg traten keine schweren Nebenwirkungen auf. Unter 10mg/kg wurde bei 2 Teilnehmern eine asymptomatische Hypomagnesiämie beobachtet. Alle Teilnehmer:innen zeigten bis Woche 13 einen Anstieg des Exon-Skippings auf median 4,26% und eine mittlere Dystrophinproduktion von 3,11% gegenüber dem Ausgangswert von 0,47%.9
Diese unerwartet geringe Exposition und die geringe Dystrophinproduktion deuteten jedoch darauf hin, dass wesentlich höhere Dosen verabreicht werden müssten, um klinisch bedeutsame Dystrophinwerte zu erreichen. Das Unternehmen stellte daraufhin die Entwicklung von PGN-EDO51 im Zusammenhang mit DMD ein.
FORWARD-53: verbesserte Muskelfunktion durch Exon-53-Skipping
WVE-N531 ist ein Exon-53-Skipping-Oligonukleotid mit einem modifizierten Rückgrat, das eine erhöhte Stabilität, Wirkeffizienz und eine bessere Verfügbarkeit im Gewebe verspricht. Die einarmige Phase-II-Studie FORWARD-53 untersuchte die Sicherheit und die funktionalen Outcomes unter WVE-N531. Li-Jung Tai von Wave Life Sciences in Cambridge, USA, stellte den Teil B der Studie vor, bei dem 11 Patient:innen im Alter von 5 bis 11 Jahren (10 davon gehfähig) alle zwei Wochen eine WVE-N531-Dosis von 10mg/kg über einen Zeitraum von 48 Wochen erhielten.
WVE-N531 war sicher und gut verträglich: Alle auftretenden behandlungsbedingten Nebenwirkungen waren leicht bis mittelschwer. Die gemessene Dystrophinexpression (Western Blot) stabilisierte sich zwischen der 24. und 48. Dosierungswoche mit einem Mittelwert von 7,8%. Zusammen erreichten 88% der Patient:innen einen durchschnittlichen Dystrophinwert von über 5% und das mittlere Exon-Skipping betrug 54%. Diese robusten Daten wurden auch durch histologische Analysen gestützt. So zeigten sich nach 48 Wochen eine Verbesserung des Muskelaufbaus, ein Übergang von Muskelregeneration zur Reifung von Muskelfasern und eine 28,6%ige Verringerung der Muskelfibrose (p<0,01).11
Klinische Verbesserungen der Muskelfunktion unter WVE-N531
Unter WVE-N531 wurden zudem klinisch bedeutsame Unterschiede im Krankheitsverlauf bei mehreren Funktionsbewertungen wie der „time to rise“ (TTR) und dem North Star Ambulatory Assessment (NSAA) erzielt. So verbesserte sich unter anderem die TTR um durchschnittlich 3,8 Sekunden gegenüber dem natürlichen Verlauf (p<0,05). Alle 11 Patient:innen sind in den Verlängerungsteil der Studie übergegangen und erhalten nun monatliche Dosen von WVE-N531.
Die durchweg positiven Daten waren Grundlage für eine neu geplante globale Bestätigungsstudie. Zudem soll 2026 ein Antrag bei der amerikanischen Food and Drug Administration (FDA) eingereicht werden, um eine beschleunigte Zulassung von WVE-N531 mit monatlicher Dosierung zu unterstützen.11
Vielversprechende erste Datenaus der INSPIRE-DUCHENNE-Studie
Eine weitere DMD-Therapieoption ist der Einsatz von Mikrodystrophinen, um die Level von funktionalem Dystrophin zu erhöhen. SGT-003 ist ein Mikrodystrophin der zweiten Generation, das die Exons 42–45 und spezifisch die nNOS-Bindedomäne beinhaltet, wodurch unter anderem eine bessere Durchblutung sichergestellt werden soll, um aktivitätsbedingte Ischämien und Muskelverletzungen zu verhindern.12 Zusätzliche Modifikationen von SGT-003 sind das SLB101-Kapsid, das in dystrophen Muskeln hochregulierte Integrine anvisiert, und eine verbesserte Ratio von vollem zu leerem Kapsid mit >80% zu <20%.13 Zusammen führen diese Anpassungen zu einer höheren Biodistribution und Expression in Skelettmuskeln und im Herzmuskel.
Hohe Mikrodystrophinexpressionslevel und Verbesserung der Muskelintegrität
Die Studie INSPIRE DUCHENNE ist eine Phase-I/II-Studie zur Sicherheit und Wirksamkeit einer einmaligen Gabe des Gentherapeutikums SGT-003 (1,0 E 14vg/kg) bei pädiatrischen DMD-Patient:innen. Prof. Dr. Kevin M. Flanigan vom Nationwide Children’s Hospital in Columbus, USA, stellte die Daten der Kohorte 1 (4 bis <7 Jahren alt, n=9) und der Kohorte 2 (7 bis <12 Jahren alt, n=6) vor.
Zum Cut-off in 3/2025 traten bei keinem der 15 Patient:innen schwerwiegende Nebenwirkungen auf. Die ersten Muskelbiopsiedaten von drei Patienten, die bereits 90 Behandlungstage abgeschlossen hatten, zeigten konstant hohe Transduktionswerte mit einem Mittelwert von 18,7 für die Vektorkopienzahl. Die Mikrodystrophinexpression lag bei 110% des normalen Dystrophinwerts und eine immunhistochemische Analyse zeigte 78% Mikrodystrophin-positive Muskelfasern (Abb.2).

Abb. 2: Die ersten Biopsiedaten pädiatrischer Patient:innen in der Studie INSPIRE DUCHENNE zeigten eine Wiederherstellung der Mikrodystrophinexpression via Western Blot unter der Gentherapie SGT-003
Außerdem wurden Verbesserungen bei mehreren Biomarkern für die Muskelintegrität beobachtet. So waren Biomarker für Muskelverletzungen signifikant um 45–60% reduziert.14 Aufgrund der positiven Daten wurde die Studie rezent erweitert und ältere Patient:innen wurden für die Kohorten 4 und 5 aufgenommen.
Update: myotone DystrophieTyp 1 (DM1)
Die DM1 ist eine autosomal-dominant vererbte Muskelkrankheit, bei der eine Triplet-Repeat-Expansion im „Dystrophia myotonica protein kinase“-Gen (DMPK) zu einer reduzierten Produktion der Myotoninproteinkinase führt.
Initiale vielversprechende Daten zu PGN-EDODM1 bei DM1
PGN-EDODM1 ist ein peptidkonjugiertes Oligonukleotid (PPMO), das auf der EDO-Peptidtechnologie basiert und im Setting der DM1 evaluiert wird. Bei DM1 führt eine pathogene Expansion von CUG-Repeats zur Bildung von Hairpins in der DMPK-mRNA, die an Spleißfaktoren wie „muscleblind-like splicing regulator 1“ (MBNL1) stabil binden. Daraus resultierende Splicingfehler werden als ursächlich für die Erkrankung angesehen.15
„Die Wirkung von PGN-EDODM1 beruht auf dessen Bindung an die CUG-Repeats und die durch sterische Blockierung erreichten erhöhten Level von freiem MBNL1. Zudem werden die Hairpinstrukturen aufgelöst, was das Proofreading wieder ermöglicht“, erklärt Prof. Dr. Hanns Lochmüller vom Children’s Hospital of Eastern Ontario in Ottawa, Kanada. Anschließend kann MBNL1 die Splicingprofile mehrerer nachgeschalteter Transkripte wiederherstellen, die eine zentrale Ursache für die DM1-Pathologie sind.16
Hohe Splicingkorrekturraten nach PGN-EDODM1-Einzeldosis
FREEDOM-DM1 ist eine placebokontrollierte Phase-I-Studie an Patient:innen mit DM1. Diese erhielten eine einzelne PGN-EDODM1-Dosis mit einer Konzentration von 5mg/kg (n=8), 10mg/kg (n=8) oder 15mg/kg (n=8).
Die initialen klinischen Daten zeigten bereits einen dosisabhängigen Anstieg der MBNL1-Konzentrationen im Muskelgewebe. Unter der höchsten Dosis von 15mg/kg wurde mit 53,7% die bisher höchste gemessene Rate an Spleißkorrekturen erreicht (Abb. 3). Zudem lagen bei 100% der Patient:innen Spleißkorrekturen vor. Dies lässt vermuten, dass zusätzliche Gaben von PGN-EDODM1 diese Werte noch verbessern können.
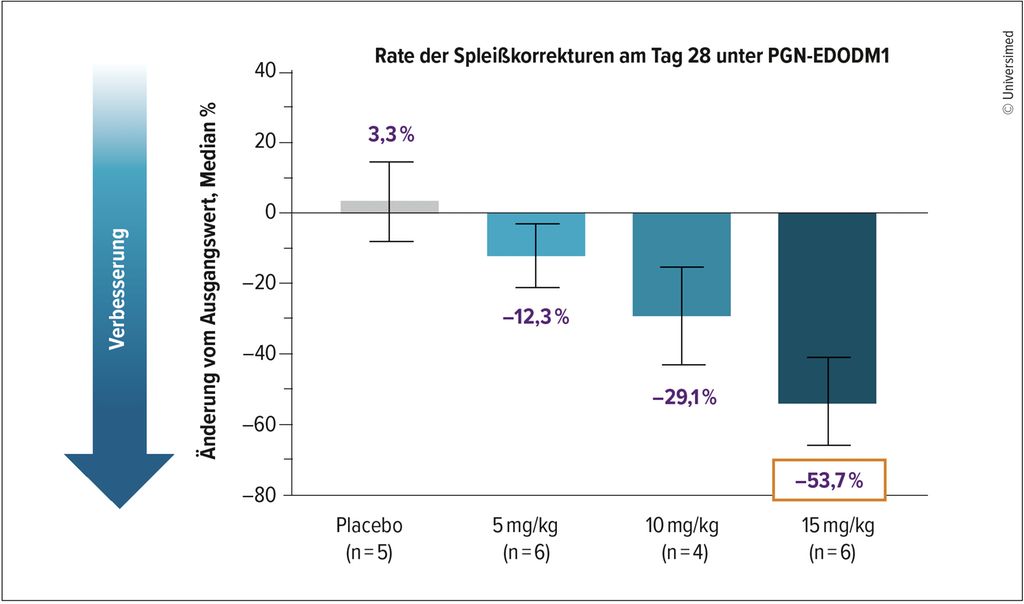
Abb. 3: Die Phase-I-Studie FREEDOM-DM1 zeigte bereits vielversprechende Ergebnisse in der Korrektur des fehlerhaften Spleißens bei Patient:innen mit myotoner Dystrophie Typ 1 am Tag 28 unter PGN-EDODM1
Des Weiteren zeichnete sich ein günstiges Sicherheitsprofil für PGN-EDODM1 ab: Es traten keine assoziierten schweren Nebenwirkungen auf.16 Auf Grundlage der vielversprechenden Daten wurde die internationale Phase-II-Studie FREEDOM2-DM1 initiiert, die die Sicherheit und den Nutzen einer Behandlung mit multiplen Dosen PGN-EDODM1 untersucht.17
Quelle:
Vorträge aus der „Clinical Trial Updates Session“ vom WMS-Kongress, 7. bis 11.10.2025, Wien
Literatur:
1 Govoni A et al.: Time is motor neuron: therapeutic window and its correlation with pathogenetic mechanisms in spinal muscular atrophy. Mol Neurobiol 2018; 55: 6307-18 2 Ratni H et al.: Discovery of risdiplam, a selective survival of motor neuron-2 ( SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem 2018; 61: 6501-17 3 Poirier A et al.: Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect 2018; 6: e00447 4 An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy (NCT03779334)5 Finkel RS et al.: Risdiplam in presymptomatic spinal muscular atrophy. N Engl J Med 2025; 393: 671-82 6 Crawford TO et al.: Continued benefit of nusinersen initiated in the presymptomatic stage of spinal muscular atrophy: 5-year update of the NURTURE study. Muscle Nerve 2023; 68(2): 157-70 7 Finkel RS et al.: DEVOTE study exploring higher dose of nusinersen in spinal muscular atrophy: study design and part A results. J Neuromuscul Dis 2023; 10: 813-23 8 Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy(NCT04089566) 9 A phase 2, open-label, multiple ascending dose study of PGN-EDO51 with a long-term extension in participants with duchenne muscular dystrophy amenable to exon 51-skipping treatment (CONNECT1-EDO51) (NCT06079736) 10 Holland A et al.: P27 three novel enhanced delivery oligonucleotide candidates for duchenne muscular dystrophy mediate high levels of exon 53, 45, and 44 skipping. Neuromuscular Disorders 2023; 33 (Suppl1): 103-4 11 An open-label phase 1b/2 study of WVE-N531 in patients with duchenne muscular dystrophy (NCT04906460) 12 Lai Y et al.: Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 2009; 119(3): 624-35 13 Vu Hong A et al.: An engineered AAV targeting integrin alpha V beta 6 presents improved myotropism across species. Nat Commun 2024; 15(1): 7965 14 A phase 1/2, multicenter, open-label study to investigate the safety, tolerability, and efficacy of a single intravenous dose of SGT-003 in males with duchenne muscular dystrophy (INSPIRE DUCHENNE) (NCT06138639) 15 Rahm L et al.: Myotonic dystrophy type 1: clinical diversity, molecular insights and therapeutic perspectives. Nat Rev Neurol 2025; 21: 623-41 16 Safety, tolerability, PK, and PD study of PGN-EDODM1 in participants with myotonic dystrophy type 1 (FREEDOM-DM1) (NCT06204809)17 A phase 2 randomized, double-blind, placebo-controlled, multiple ascending dose study of PGN-EDODM1 in adult participants with myotonic dystrophy type 1 (FREEDOM2-DM1) (NCT06667453)
Das könnte Sie auch interessieren:
Künstliche Intelligenz in der Neuroradiologie: Chancen, Herausforderungen und klinische Anwendungen
Künstliche Intelligenz bietet vielfältige Chancen für die Neuroradiologie. Richtig eingesetzt, kann sie die Effizienz und Genauigkeit in der Radiologie verbessern. Doch es gibt ...

Hypersomnolenz im Fokus
5–10% der Allgemeinbevölkerung sind von Wachstörungen betroffen. Diese haben ein breites Spektrum und umfassen häufige Störungen wie Müdigkeit, Fatigue oder Aufmerksamkeitsstörungen. ...
ECTRIMS: State of the Art – Serumbiomarker für PIRA
Obwohl aktuelle Therapien die akut-entzündliche Aktivität der Multiplen Sklerose (MS) meist sehr gut behandeln, kommt es nicht selten zu einer schubunabhängigen Krankheitsprogression ( ...