
Kutane Lymphome: zielführende Therapie trotz klinischer Mimikry
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Kutane Lymphome treten mit einer Inzidenz von 1:100000 relativ selten auf. Es lohnt sich, sich mit ihnen zu befassen, da sie zum Teil schwer erkennbar, zu einem erheblichen Anteil progredient und dann mit hoher Mortalität verbunden sind. Diese Mycosis fungoides nimmt besonders in Frühstadien sehr vielgestaltige Ausprägungen an.
Keypoints
-
Die frühe Mycosis fungoides (MF) ist nicht nur klinisch, sondern auch häufig histologisch schwer zu diagnostizieren.
-
Aufmerksamkeit ist geboten bei fehlendem Ansprechen oder Progression einer AD oder Psoriasis unter Biologikatherapie, weil tatsächlich eine MF vorliegen kann.
-
Der Einfluss von Dupilumab auf das Auftreten und die Progression einer MF bleibt noch genauer zu klären.
Definitionsgemäss treten kutane Lymphome primär in der Haut auf, ohne extrakutane Manifestation zum Zeitpunkt der Diagnose, berichtete Dr. med. Matthias Steinhoff, Berlin, anlässlich der DERM Frankenthal. Es sei wichtig, den sekundären Hautbefall eines extrakutanen Lymphoms auszuschliessen. Verglichen mit nodalen Lymphomen zeigen kutane einen deutlich besseren Verlauf.
Die 2019 erschienene WHO-EORTC- Klassifikation unterscheidet zwischen den indolenten und den aggressiven kutanen T-Zell-Lymphomen (CTCL) mit jeweils verschiedenen Subtypen.1 Bei rund 45% der CTCL handelt es sich um eine Mycosis fungoides (MF) und ihre Subtypen, einschliesslich des seltenen Sézary-Syndroms. Etwa ein Viertel stellen die B-Zell-Lymphome, ein weiteres Fünftel die CD30-positiven Lymphoproliferationen. Kennt man die genannten Entitäten, dann kann man in der Praxis 90% der kutanen Lymphome diagnostizieren, nur ein kleiner Teil entfällt auf seltenere Formen, so Steinhoff. Eine WHO-Klassifikation der kutanen Lymphome von 2024 ergänzt die aktuellen Daten.2
Das Chamäleon unter den Dermatosen: die Mycosis fungoides
Eine rasche Diagnose der MF wird durch ihre besonders in Frühstadien sehr vielfältige Ausprägung erschwert. Die Erkrankung präsentiert sich eher ausnahmsweise typisch, also mit Patches in UV-geschützten Arealen, mit Plaques entlang der Hautspannungslinien oder mit Tumoren. Tatsächlich kann die MF klinisch mehr als 50 unterschiedliche dermatologische Erkrankungen imitieren,3 darunter ekzematöse, psoriasiforme und papulöse Dermatosen. Gefunden werden aber auch Hyperkeratosen und Poikilodermien sowie hyper- oder hypopigmentierte und elastolytische Formen.
Auch die histologischen Befunde sind häufig unspezifisch: Es finden sich ein oberflächliches perivaskuläres Infiltrat oder eine Dermatitis, kaum jedoch Epidermotropismus und wenig atypische Zellen. Die klassische MF präsentiert sich mit atypischen Lymphozyten, die sich entlang der Basalzellenreihe zu Pautrier-Mikroabszessen formieren, sowie einem dominanten T-Zell-Klon. Der EORTC-Gruppe «Kutane Lymphome» zufolge wird die MF im Schnitt mit einer Verzögerung von median 36 Monaten diagnostiziert.
Schlechtere Prognose durch verzögerte MF-Diagnose?
Wenn Biologika zum Einsatz kommen, gewinnt eine verzögerte Diagnose an Bedeutung, warnte Steinhoff. In einem bereits vor 10 Jahren publizierten Fall hat eine Patientin wegen ihrer Psoriasis den TNFa-Inhibitor Infliximab erhalten, kurz darauf wurde massive MF mit grosszelliger Transformation diagnostiziert und die Patientin ist daraufhin verstorben.4 Bei zwei anderen Patienten mit vermuteter Psoriasis trat nach dem Einsatz des monoklonalen Interleukin(IL)-17-Antikörpers Secukinumab eine MF auf.5
Ein weiterer Patient erhielt aufgrund seiner vermeintlichen atopischen Dermatitis (AD) den monoklonalen Antikörper Dupilumab, der den IL-4- und IL-13-Signalweg blockiert. Das Krankheitsbild verschlechterte sich daraufhin deutlich und man diagnostizierte auch hier schliesslich MF.6 In der Literatur der letzten 4 Jahre findet man zunehmend Berichte über nicht erkannte MF-Patient:innen, bei denen es unter einem Therapieversuch mit Dupilumab zu einer Progression kam.7 In einem Review wurde bereits ein erhöhtes Risiko für kutane T-Zell-Lymphome nach Dupilumab-Anwendung vermutet.8
In einer Studie an 371 Patient:innen mit atopischer Dermatitis (AD) sprachen 325 sehr gut auf Dupilumab an, 46 jedoch nicht. Von ihnen wurden 35 Patient:innen reevaluiert: Bei 16 davon bestätigte sich die AD-Diagnose, bei 19 lag eine MF vor.9
Ausserdem zeigte eine auf den Daten eines Patient:innenregisters beruhende Arbeit zur AD, dass die Kohorte unter Dupilumab-Therapie ein vierfach höheres Risiko für kutane T-Zell-Lymphome hatte als jene ohne Dupilumab-Therapie (Odds-Ratio [OR] 4,1). Die CTCL traten meist innerhalb eines Jahres unter Dupilumab auf. Andere lymphoproliferative Erkrankungen oder solide Tumoren der Haut wurden nicht vermehrt beobachtet.10
Eine Pharmakovigilanzstudie zu verschiedenen Biologika, die sich auf eine halbe Million registrierter unerwünschter Ereignisse stützte, ermittelte eine OR von 8,8 für das Auftreten kutaner Lymphome unter Dupilumab. Dies war die höchste verglichen mit einem IL-23-Inhibitor (OR 4), einem IL-17-Inhibitor (OR 3,3), einem IL-12/23-Inhibitor (OR 2,2) und einem TNFa-Blocker (OR 5,8).11
Insgesamt sei das Bild zur Progression der MF unter Dupilumab widersprüchlich. Dies beruht zum Teil auf einer Fehldiagnose, erläuterte Steinhoff. Es gebe jedoch sowohl Daten, die eine Besserung zeigten, als auch solche, bei denen das nicht der Fall sei. Zu klären bleibt, welchen Einfluss die IL-4-/IL-13-Blockade auf Tumorzellen und das Tumormikromilieu habe. Möglicherweise fördern erhöhte IL-13-Spiegel im Gewebe die MF-Progression. Vor der Einleitung einer Biologikatherapie, insbesondere mit Dupilumab, empfahl Steinhoff deshalb eine exakte Diagnosestellung, bei atypischer AD oder Psoriasis auch eine histologische Abklärung. Bleibt ein Ansprechen aus oder kommt es zum Progress, muss eine MF ausgeschlossen werden.
Neuere Optionen: niedrig dosierte Ganzhautbestrahlung und Biologika
Zunächst ist für die Therapieauswahl zwischen frühen CTCL-Stadien mit Patches und Plaques und fortgeschrittenen Stadien mit Tumoren zu unterscheiden, der Lymphknotenstatus sowie die Blut-Organ-Beteiligung sind abzuklären. Die Prognose der MF lasse sich anhand des Hautbefalls bereits gut abschätzen, so Steinhoff weiter. Eine sehr gute Prognose besteht in frühen Stadien. Wenn Tumoren oder Erythrodermie voriegen, sinkt das mediane Gesamtüberleben jenseits des Tumorstadiums IIB aber auf unter 5 Jahre.12
Bei etwa einem Drittel aller Betroffenen entwickelt sich die Erkrankung progredient. Zu den dann möglichen hautgerichteten Therapien gehören lokale Kortikosteroide plus UV-Therapie sowie Chlormethin-Gel. Systemisch kommen pegyliertes Interferon alpha oder Bexaroten infrage, extrakorporale Photopherese (ECP) ab Stadium III oder Bestrahlung mit schnellen Elektronen. Als zielgerichtete Therapien eignen sich Mogamulizumab, Brentuximab-Vedotin, Alemtuzumab bzw. als zytoreduktive Therapeutika Gemcitabin, MTX, Doxorubicin und CHOP, ausserdem die allogne Stammzelltransplantation, wie auch in der S2k-Leitlinie «Kutane Lymphome» nachzulesen ist, fasste Steinhoff zusammen.13
Das seit 8 Jahren zugelassene Chlormethin-Gel erreicht ein Ansprechen von über 50% in frühen Erkrankungsstadien bis IIa.14 Die Nebenwirkungen lassen sich etwas senken, indem man es alternierend mit topischen Kortikosteroiden verabreicht.15
Einen grossen Stellenwert hat auch die Ganzhaut-Elektronenbestrahlung, die nicht mehr hoch dosiert (30–50Gy), sondern mit niedrigen Dosen (12G) erfolgt, so Steinhoff. Sie ist auch bei fortgeschrittenen Stadien wirkungsvoll.16 In einer aktuellen Arbeit präsentiert die EORTC «Kutane Lymphome» Behandlungspfade sowohl für T-Zell-Lymphome als auch für B-Zell-Lymphome.17
CTCL-Zweitlinientherapie: Mogamulizumab und Brentuximab-Vedotin
Bei Erkrankten mit MF oder Sézary-Syndrom kommt es zu einer massiven Überexpression des Chemokin-Rezeptors 4 (CCR4). Das Zytokin gehört (mit CCL17/CCL22) zur «Achse des Bösen», denn es sei letztendlich verantwortlich für das «Homing» maligner T-Zellen in die Haut, ein TH2-dominiertes Zytokinmilieu, für die Hemmung zytotoxischer T-Zellen und attrahiere immunsuppressive Makrophagen, machte Steinhoff deutlich. Insgesamt begünstige dies das Überleben der Tumorzellen (Abb. 1).18
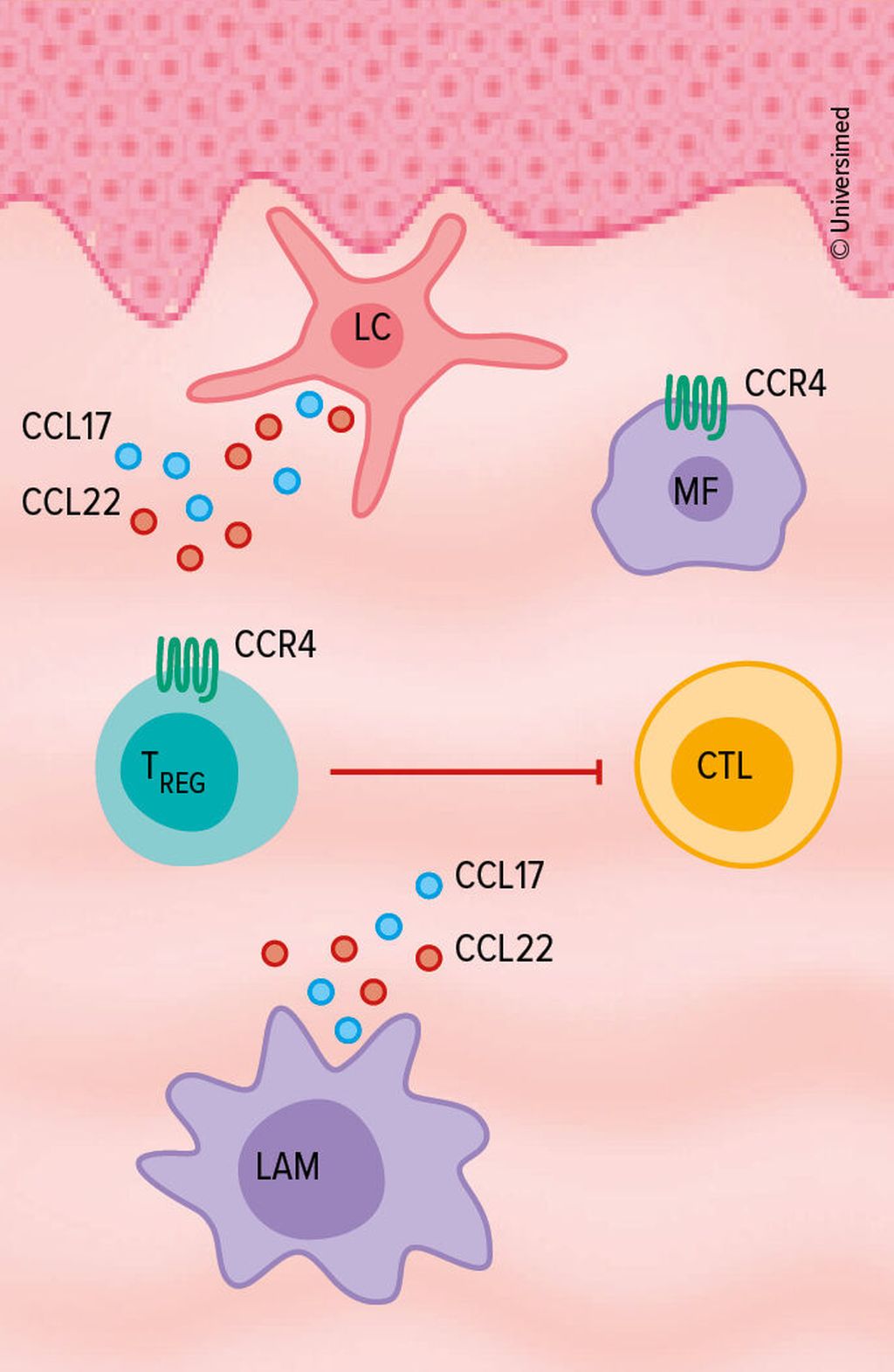
Abb. 1: Die CCR4-Überexpression bei MF oder Sézary-Syndrom begünstigt das Überleben von Tumorzellen (modifiziert nach Wilcox RA 2015)18
Bei Patient:innen mit zuvor frustraner systemischer Therapie kann Mogamulizumab eingesetzt werden. Indem man so den CCR4 blockiert, depletiert man die CCR4-positive Tumorzelle und gleichzeitig die immunsuppressive T-regulatorische Zelle. Dass dieses Konzept auch in der Praxis funktioniert, hat die Mogamulizumab-Zulassungsstudie gezeigt, in der das Biologikum mit Vorinostat verglichen wurde, einem Inhibitor, der seit circa 20 Jahren in den USA zugelassen ist, in Deutschland oder in Europa jedoch nicht. Sowohl hinsichtlich des medianen progressionsfreien Überlebens (7,7 vs. 3 Monate) als auch hinsichtlich des Skindex-29, also der gesamten Symptomatik, ergaben sich signifikante Vorteile unter Mogamulizumab.19 In einer Post-hoc-Analyse zeigte sich, dass die Patient:innen mit Blutbeteiligung und schlechter Prognose sehr von Mogamulizumab profitieren, auch jene mit Sézary-Syndrom.20
Der Mogamulizumab-assoziierte Rash, eine überschiessende Immunreaktion, tritt in 15 bis 20% der Fälle auf. Es sei schwierig, ihn von einem CTCL-Progress abzugrenzen, dies sei jedoch wichtig, weil dieser Rash mit einem besseren Ansprechen korreliere, appellierte Steinhoff.21
Ein zweiter Antikörper, Brentuximab-Vedotin, wird bei CD30-positiven grosszelligen Lymphomen oder auch bei MF eingesetzt und ist ein Antikörper-Wirkstoff-Konjugat aus einem gegen CD30 gerichteten rekombinanten monoklonalen AK und dem Antimikrotubuli-Wirkstoff Monomethylauristatin E (MMAE). Dieser wird internalisiert, zerstört Mikrotubuli und Spindelapparat und es kommt zum Zellzyklusarrest und zur Apoptose (Abb.2). Hierfür wird nicht viel CD30 benötigt. Die MF ist typischerweise nicht CD30-positiv, doch sei es wichtig, im histologischen Bericht «in-label» zu bleiben und zumindest 1 bis 2 Zellen als CD30-positiv zu beschreiben, führte Steinhoff aus.22

Abb. 2: Brentuximab-Vedotin-CD30-Antikörper. Der Einsatz führt zur Zerstörung des Spindelapparates, zum Zellzyklusarrest und schliesslich zur Apoptose (modifiziert nach Siddiqi T et al. 2014)22
In einer Studie an 100 Teilnehmenden mit MF und 31 mit CD30-positivem großzelligem Lymphom wurde Brentuximab-Vedotin mit Methotrexat oder Bexaroten verglichen. Sie zeigte mit 65% vs. 20% kompletten Remissionen ein überlegenes Ansprechen auf Brentuximab-Vedotin. Ausserdem war das mediane progressionsfreie Überleben mit 16,7 Monaten vs. 3,5 Monate deutlich überlegen.23
Quelle:
«Update Kutane Lymphome», Vortrag von Dr. med. Matthias Steinhoff, Berlin, im Rahmen des «onkoderm-Symposiums», 21. März 2025, DERM 2025, Frankenthal
Literatur:
1 Willemze R et al.: Blood 2019; 133: 1703-14 2 Melchers S et al.: JGGG 2024; doi:10.1111/ddg.15361 3 Wobser M et al.: World J Dermatol 2015; 4: 135-44 4 Suga H et al.: Acta Derm Venereol 2014; 94: 233-4 5 Yoo J et al.: Clin Exp Dermatol 2019; 44: 414-7 6 Chiba T et al.: Acta Derm Venereol 2019; doi:10.2340/00015555-3208 7 Espinosa ML et al. J Am Acad Dermatol. 2020; 83: 197-9 8 Mandel J et al.: Dermatol Ther 2024; doi:10.1155/2024/9924306 9 Kook H et al.: J Eur Acad Dermatol Venereol 2024; doi:10.1111/jdv.20053 10 Hassan I et al.: J Am Acad Dermatol 2024; 91: 255-8 11 Lavin L et al.: J Investig Dermatol 2024; 145: 211-4.e1 12 Agar NS et al.: J Clin Oncol 2010; 28: 4730-9 13 Dippel E et al.: JDDG 2022; 20: 537-55 14 Lessin SR et al.: JAMA Dermatol 2013; 149: 25-32 15 Assaf C et al.: JDDG 2022; doi:101111/ddg.14688 16 Morris S et al.: Int J Radiat Oncol Biol Phys. 2017; 99: 627-33 17 Elsayad K et al.: Eur J Cancer 2024; doi:10.1016/j.ejca.2024.115064 18 Wilcox RA: Blood 2015; 125(12): 1847-8 19 Kim YH et al.: Lancet Oncol 2018; 19: 1192-204 20 Cowan RA et al.: J Eur Acad Dermatol Venereol 2021; 35; 2225-38 21 Assaf C et al.: JDDG 2024; doi:10.1111/ddg.15639 22 Siddiqi T et al.: Pharmgenomics Pers Med 2014; 7: 79-85 23 Horwitz SM et al.: SM Blood Adv 2021; 6: 5098-106
Das könnte Sie auch interessieren:

Frauenhaut wird anders krank, Männerhaut auch
Asthma wird bei Mädchen später diagnostiziert als bei Jungen, Parkinson und Harnblasenkarzinom bei Frauen später als bei Männern. Auch ein Myokardinfarkt wird bei Frauen immer noch ...

Im Fokus: VEXAS-Syndrom
Es tritt vermutlich viel häufiger auf als bisher angenommen: das autoinflammatorische VEXAS-Syndrom. Die chronisch progressiv verlaufende Erkrankung wird zumeist sehr spät diagnostiziert ...
Kombinationstherapie mit plättchenreichem Plasma und Hyaluronsäure
Hochwertiges autologes plättchenreiches Plasma (PRP) verfügt von Natur aus über einen komplex zusammengesetzten Cocktail aus zahlreichen bioaktiven Substanzen. Gegenüber dem Vollblut ...